Ich lese (vor allem bei Twitter) immer wieder als Begründung für den Wunsch nach Beibehaltung der Corona-Maßnahmen (die ja eigentlich nicht-pharmazeutische Interventionen (NPI) heißen), dass man Angst oder Sorge vor Long Covid auch als geimpfter Mensch habe. Und ich frage mich dann (auch immer wieder), ob das eigentlich eine rationale Angst oder Sorge ist.
Disclaimer: Ich kenne eine geimpfte Person, die sich mit Omikron infiziert hat und nun Beschwerden hat, die ich unter Long Covid subsumieren würde. Von daher, abwegig ist der Gedanke Long Covid zu bekommen nicht.
Disclaimer 2: Ich hantiere hier mit absoluten Risiken, nicht mit relativen.
Ich hatte mir schon vor einiger Zeit überlegt, dass das sinnvollste für mich persönlich wäre, das Erkrankungsrisiko mit den Erkrankungen zu vergleichen, mit denen ich es tagtäglich zu tun habe und bei denen ich sicher bin, dass ich sie nicht haben will, weil sie einen relevanten Impact auf mein tägliches Leben hätten.
Hier ist also meine Recherche, wenn ihr sie lesen wollt nur zu, sonst lest lieber zum Beispiel diesen Beitrag über das Demenz-Risiko von Fußballprofis Link.
Das Long Covid-Risiko
Das Schwierigste war es, sich auf ein Long Covid-Erkrankungsrisiko festzulegen, da hier – bekanntermaßen – ja sehr heterogene Häufigkeitsangaben auftauchen. Auch ist Long Covid nicht gleich Long Covid (hier gibt es eine was die Studien betrifft leicht veraltete, aber in den Grundzügen immer noch aktuelle Beitragsserie dazu (Link)) und ein großer Teil der Betroffenen hat für einen gewissen Zeitraum nach der Infektion noch Beschwerden und ein deutlich kleinerer sehr viel länger bis dauerhaft. Auch weiß ich aus eigener Erfahrung, dass ich nach meiner COVID-Infektion Ende 2020 mehr als vier Wochen nicht richtig riechen und schmecken konnte (was formal Long Covid-Kriterien erfüllt hätte) und noch ewig einen fiesen Reizhusten hatte (den ich aber nach jedem Infekt ewig lang habe und irgendwann mit Cortison-Spray beende).
Was einem zudem auch niemand bislang verlässlich sagen kann ist, wie es sich mit dem Long Covid-Risiko nach den zu erwartenden Reinfektion 2-x verhält, einfach weil es recht wenig Reinfektionen gibt, mit denen man das untersuchen kann. Es gibt aber ein paar Hinweise, die man verwenden kann: Das Long Covid-Risiko scheint stark von der Krankheitsschwere abzuhängen, je schwerer der Verlauf war, umso wahrscheinlicher sind länger anhaltende Beschwerden. Und Reinfektionen verlaufen bei den meisten Betroffenen, gerade wenn sie geimpft sind, deutlich leichter als Primärinfektionen bei Ungeimpften. Prinzipiell kennen wir mittlerweile neben der initialen Krankheitsschwere auch weitere Hauptrisikofaktoren für Long Covid: Weibliches Geschlecht, komorbide psychiatrische Erkrankungen, erhöhter ANA-Titer als Ausdruck eines autoimmunologischen Geschehens, hoher BMI/Diabetes mellitus, EBV/CMV-Reaktivierung (z.B.: Su Y, Yuan D, Chen DG, et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell. 2022;185(5):881-895.e20. doi:10.1016/j.cell.2022.01.014 oder Crook, H., Raza, S., Nowell, J., Young, M., & Edison, P. (2021). Long covid—mechanisms, risk factors, and management. iBMJ, n1648. https://doi.org/10.1136/bmj.n1648).
Nach vielem hin-und-her-Überlegen hab ich mich für dieses israelische Preprint entschieden, einfach weil es relativ aktuell ist, weil es relativ „großzügig“ mit der Long Covid-Diagnose umgeht und dementsprechend hohe Prävalenzen bietet (und so ein worst-case-Szenario darstellen könnte) und weil es geimpfte und ungeimpfte Probanden mit COVID-19 mit einer Kontrollgruppe ohne COVID-19 vergleicht:
Kuodi P, Gorelik Y, Zayyad H, et al. Association between Vaccination Status and Reported Incidence of Post-Acute COVID-19 Symptoms in Israel: A Cross-Sectional Study of Patients Tested between March 2020 and November 2021. Epidemiology; 2022. doi:10.1101/2022.01.05.22268800
Von den COVID-Infizierten ohne Impfung gaben 35% in der Studie anhaltende Beschwerden nach der Infektion an. Am häufigsten wurde eine anhaltende Müdigkeit, also Fatigue angegeben (26%), diese wurde aber auch von 18% der Nicht-Infizierten berichtet. 22% der Infizierten und 16% der Nicht-Infizierten berichteten von Kopfschmerzen, 16% (8% Kontrollgruppe) von einem Schwächegefühl der Beine, 11% (4%) von Muskelschmerzen, 12% (6%) von Konzentrationsstörungen, 13% (4%) von Haarausfall, 10% (8%) von Schlafstörungen, 8% (6%) von Husten und 8% (3%) von Kurzatmigkeit.
Das bedeutet, dass das absolute Risiko Long Covid-Beschwerden im Vergleich zu einer nicht-infizierten Kontrollgruppe zu entwickeln, ungefähr 9% mehr nach Infektion auftraten, als in der Normalbevölkerung eh vorhanden. Bei vollständig Geimpften mit einer COVID-Infektion (im Studienzeitraum waren das noch zwei Impfungen) waren die Symptome zwischen 54% und 82% seltener (CAVE, das ist jetzt ein relatives Risiko) als bei Ungeimpften und – merkwürdigerweise – bei allen Symptomen bis auf Muskelschmerzen, anhaltendem Husten und Kurzatmigkeit sogar seltener als bei Nicht-Infizierten.
aus: Kuodi P, Gorelik Y, Zayyad H, et al. Association between Vaccination Status and Reported Incidence of Post-Acute COVID-19 Symptoms in Israel: A Cross-Sectional Study of Patients Tested between March 2020 and November 2021. Epidemiology; 2022. doi:10.1101/2022.01.05.22268800, direkter Link zur Quelle.
Über den Daumen gepeilt muss ich also als geimpfter, übergewichtiger Mann mittleren Alters mit einem Long Covid-Risiko von so 5% nach einer COVID-Infektion rechnen.
Die Vergleichs-Erkrankungen
Wie ich oben schon geschrieben habe, habe ich mal die neurologischen Erkrankungen mit denen ich es am häufigsten zu tun habe herausgesucht, die einen relevanten Impact auf mein tägliches Leben hätten. Ich habe zusätzlich noch den Diabetes mellitus hinzugenommen, da ich als kleiner Dicker da sicherlich ein relevantes Risiko für habe und weil ein Diabetes im Vergleich z.B. zu Bluthochdruck deutlich mehr Lebensstilveränderungen erfordert.
Ich habe hier – wenn es ging – nach den Lebenszeitprävalenzen gesucht, also dem Risiko irgendwann im Leben diese Erkrankung zu bekommen. Der Vergleich mit Long Covid haut dann nicht hin, wenn man jetzt mehrfach im Leben Long Covid bekommen sollte. Aber ob das überhaupt ein realistisches Szenario ist, weiß wirklich niemand (siehe oben).
Schlaganfall
Die Lebenszeitprävalenz einen Schlaganfall zu erleiden, liegt ungefähr bei 25%:
The GBD 2016 Lifetime Risk of Stroke Collaborators. Global, Regional, and Country-Specific Lifetime Risks of Stroke, 1990 and 2016. N Engl J Med. 2018;379(25):2429-2437. doi:10.1056/NEJMoa1804492
Diabetes mellitus
Das Risiko an einem Diabetes mellitus zu erkranken hängt extrem von den Lebensumständen, dem Körpergewicht, Geschlecht, Alter und der ethnischen Zugehörigkeit ab, liegt aber zwischen 20% und 40% Lebenszeitprävalenz.
Narayan KMV. Lifetime Risk for Diabetes Mellitus in the United States. JAMA. 2003;290(14):1884. doi:10.1001/jama.290.14.1884
Epilepsie
Die Lebenszeitprävalenz für eine Epilepsie liegt nach einer aktuellen Studie bei 7,6/1.000 Personen:
Fiest KM, Sauro KM, Wiebe S, Patten SB. Prevalence and incidence of epilepsy. Published online 2016:8.
Hirntumore
4-5/1.000 beträgt das Lebenszeit-Erkrankungsrisiko für einen bösartigen hirneigenen Tumor, also ein Gliom oder ein Astrozytom. Für Krebserkrankungen ganz allgemein gibt es Daten zur 10-Jahres-Prävalenz (also, wie viele Menschen innerhalb von 10 Jahren an einem bösartigen Tumor erkranken). Diese liegt bei Männern bei 0,1% und bei Frauen bei 0,04% (Link), insgesamt leben in Deutschland 1,9% der Bevölkerung mit einer Krebsdiagnose (Link).
Rice T, Lachance DH, Molinaro AM, et al. Understanding inherited genetic risk of adult glioma – a review. Neuro-Oncology Practice. 2016;3(1):10-16. doi:10.1093/nop/npv026
Multiple Sklerose
Bei der MS wissen wir ja seit kurzem, dass eine durchgemachte EBV-Infektion kausal mit der Erkrankung zusammenhängt und nach EBV das MS-Erkrankungsrisiko bei ca. 1:900 liegt und nach durchgemachter infektiöser Mononukleose (also dem Pfeiffer’schen Drüsenfieber) bei 1:240. Da die Durchseuchung bei EBV bei 95% liegt, korreliert es mit der Prävalenzangabe aus anderen Studien von 0,1% Lebenszeitrisiko. Das ist natürlich deutlich seltener als Long Covid, ist (für mich) aber der Prototyp für eine lebenslang vorhandene, nicht kausal heilbare Erkrankung.
Bjornevik K, Cortese M, Healy BC, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. Published online January 21, 2022:1-10. doi:10.1126/science.abj8222
Wallin MT, Culpepper WJ, Nichols E, et al. Global, regional, and national burden of multiple sclerosis 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. The Lancet Neurology. 2019;18(3):269-285. doi:10.1016/S1474-4422(18)30443-5
Neurodegenerative Erkrankungen
Diese laufen für mich ein wenig außer Konkurrenz, da sie ja vor allem im höheren Lebensalter auftreten. Dennoch liegt die Lebenszeitprävalenz für eine Alzheimer-Erkrankung bei US-Bürgern zwischen 11,6% und 21,1% (Link) oder bei älteren Daten aus der Framingham Heart Study ab einem Lebensalter von 65 Jahren zwischen 25,5 % bis 28,1% Rest-Lebenszeitprävalenz.
Seshadri S, Wolf PA, Beiser A, et al. Lifetime risk of dementia and Alzheimer’s disease: The impact of mortality on risk estimates in the Framingham Study. Neurology. 1997;49(6):1498-1504. doi:10.1212/WNL.49.6.1498
Das idiopathische Parkinson-Syndrom ist seltener, hier liegt die Lebenszeitprävalenz zwischen 3,7% bei Frauen und 4,4% bei Männern, zumindest nach den Daten dieser auch schon 20 Jahre alten Studie:
Elbaz A, Bower JH, Maraganore DM, et al. Risk tables for parkinsonism and Parkinson’s disease. Journal of Clinical Epidemiology. 2002;55(1):25-31. doi:10.1016/S0895-4356(01)00425-5
Fazit
In der Welt der neurologischen Erkrankungen lautet das Ergebnis der Recherche für mich also: Es ist wahrscheinlicher, dass ich an einem Schlaganfall, einem Diabetes mellitus oder einer neurodegenerative Erkrankung im Alter erkranke als an Long Covid. Anders herum ist eine MS-Diagnose, eine Epilepsie und ein hirneigener bösartiger Tumor unwahrscheinlicher als Long Covid. Mein ganz persönliches Fazit ist, dass ich keine Sorge und keine Angst vor Long Covid habe, aber auch nicht aus allen Wolken fallen werde, wenn ich daran erkranke. Da ich aber mit den anderen hier zitierten Erkrankungsrisiken auch vor der COVID-19-Ära schon umgegangen bin, werde ich mich wegen des Long Covid-Risikos in meinem Privat- und Sozialleben nicht einschränken. Ganz im Gegenteil.
Das ist aber nur mein Fazit, das muss nicht eures sein. Zudem bin ich auch nicht der Typ, der große Teile seines Monatseinkommens für Versicherungen und Absicherungen zurücklegt, sondern da eher mit einem „Rumpf-Programm“ arbeitet. Außerdem kann es gut sein, dass die Berechnung, die ich für mich aufgemacht habe, am Ende „nicht richtig“ ist, aber am Ende machen für mich persönlich ein paar Prozent mehr oder weniger nicht den den Unterschied.
Es gab bei Twitter – mal wieder – etwas Streit. Es ging – mal wieder – um das Thema COVID-Impfung und das Myokarditis-Risiko durch die Impfung und durch die Infektion und darum, welche Konstellation risikoreicher ist:
Klar, der Twitter-User narkosedoc polarisiert, klar, die echten Impfgegner wird man eh kaum umstimmen, aber insgesamt besteht bei dem Thema so viel Halbwissen bei so viel starker Meinung, dass es Zeit ist, sich mit Myokarditiden ohne und mit COVID, sowie nach COVID-Impfung noch mal intensiver zu beschäftigen.
Man muss sich aber – wenn man das tut – folgendes vor Auge halten: Bei pubmed finden sich Stand heute (15.02.2022) 1.257 Einträge zum Thema myocarditis + covid (Link) und 773 zum Thema myocarditis + vaccine (Link), zuzüglich unzähliger Preprints die durch Twitter geistern, Gesundheits-Survey-Daten von Behörden verschiedener Länder usw. Das führt unweigerlich zu cherrypicking, weil man immer nur einen kleinen, selektiven Teil der zur Verfügung stehenden Datenflut überblicken und benutzen kann.
Es lohnt sich, sich erst einmal mit dem Thema Myokarditis zu beschäftigen, so wie man das in der Prä-Corona-Ära getan hat, um einen generellen Überblick über das Krankheitsbild zu erlangen.
Was man bis Anfang 2020 zum Thema Myokarditis wusste: Myokarditis ohne COVID-Infektion
Wer sich zu diesem Thema – das der klassischen Myokarditis – etwas belesen will (abseits von dem, was wir hier schreiben), kann dies zum Beispiel wunderbar mit diesem Reviews tun, welches es als open access-Artikel gibt: Der Arbeit von Tschöpe et al.: Link.
Eine kleine Begriffsklärung
Als Myokarditis bezeichnet man eine Entzündung des Herzmuskels. Ganz oft ist aber auch das Bindegewebe um das Herz betroffen, dann spricht man von einer Perikarditis. Meistens sind aber Bindegewebe und Herzmuskel entzündet, so dass die korrekte Bezeichnung Perimyokarditis lauten würde. Im wirklichen Leben – und auch den hier zitierten Studien – gehen die Begrifflichkeiten oft durcheinander, teilweise werden sie synonym benutzt, teilweise auch Trennungen zwischen Peri- und Myokarditis gezogen.
Krankheitsdefinition und Ätiologie der Myokarditis
Häufigste Ursache in den westlichen Industrienationen für eine Myokarditis sind Virusinfektionen, sowohl direkt während der akuten Erkrankung als Infektionsmanifestation, als auch nach einer viralen Erkrankung als immunologisch vermitteltes Phänomen. Die häufigsten Erreger der Virus-Myokarditis haben sich in den letzten Jahren verändert. Klassischerweise waren es verschiedene Adeno- und Enteroviren (vor allem Coxsackie-Viren), die als Myokarditis-Verursacher galten. In den letzten Jahren, bzw. dem letzten Jahrzehnt, wurden dann zunehmend häufiger Parvovirus B-19 (Link Wikipedia), der Erreger der Ringelröteln und die Viren der Herpesgruppe, vor allem EBV, HHV6 und CMV als ursächlich für Myokarditiden beschrieben. Dazu kommen – je nach Serotyp und Schwere der Saison – das Influenza-Virus und zwei klassische Viren, die mit Gefäßentzündungen, also Vaskulitiden assoziiert sind: Hepatitis C-Virus und HIV.
Prinzipiell – und das ist in Entwicklungsländern zum Beispiel häufig so – können auch sehr viele Bakterien und auch Parasiten wie Clamydien, der Diphtherie-Erreger, TBC, Staphylokokken, Streptokokken, Pilze, Toxoplasmen usw. eine Myokarditis verursachen, zudem nahezu alle Autoimmunerkrankungen und verschiedene Toxine. Auch hier sind die Stoffe, die eine Vaskulitis verursachen können, besonders häufig beteiligt: Zum Beispiel Kokain und Amphetamine, aber auch Chemotherapeutika und Alkohol. Ebenfalls sind Myokarditiden nach Impfungen beschrieben, vor allem nach der Pocken-Impfung, aber auch nach Influenza- und Tetanus-Impfungen.
Blackbox Myokarditis-Epidemiologie
Schwierig wird es bei der Epidemiologie. Die Myokarditis ist vermutlich stark unterdiagnostiziert, was vor allem an der oft unspektakulären und wenig eindeutigen klinischen Präsentation liegt. Auch wurde – vor allem in der Prä-Kardio-MRT-Ära – die einzige wirklich verlässliche diagnostische Prozedur, die Endomyokardbiopsie auf Grund der Sorge vor Komplikationen nur relativ selten durchgeführt.
Also stochert man im Nebel und vermutete 2013 in den westlichen Industrienationen eine Prävalenz von 22/100.000 pro Jahr mit deutlich mehr betroffenen Männern als Frauen und mit einem besonderen Peak bei jungen Männern (Kindermann et al.). Daten aus Deutschland gibt es nicht, dafür (wie immer) britische epidemiologische Daten. Hier findet sich eine Zusammenfassung: Link. Aber auch die britischen Daten sind relativ überschaubar. Die verschiedenen Paper zur Myokarditis verweisen am Ende immer wieder auf diesen Abstract (Link), in dem NHS-Daten, welche in über 20 Jahren von 1998 bis 2017 gesammelt wurden, vorgestellt werden. In dem Abstract geht es um Krankenhauseinweisungen auf Grund einer Myokarditis, nicht um symptomatische Fälle. Die Autoren berichten über den Beobachtungszeitraum von einer Zunahme der Häufigkeit einer Aufnahme mit einer Myokarditis ins Krankenhaus um 88%. Zwei Drittel der Patienten waren männlich, das mittlere Erkrankungsalter lag bei 33 Jahren bei Männern und 46 Jahren bei Frauen. Es findet sich in dem Abstract eine kleine Grafik, in der die Krankenhausaufnahmen nach Altersgruppen aufgeschlüsselt wurden. Tabellarisch übertragen sieht das in etwa so aus (die Auflösung der Grafik ist so schlecht, dass ich sicherheitshalber gerundet habe):
Alter (Jahre)
Krankenhausaufnahmen / Jahr
0 – 4
40
5 – 9
2
10 – 14
5
15 – 19
130
20 – 24
185
25 – 29
150
30 – 34
140
35 – 40
120
40 – 44
110
45 – 49
110
50 – 54
100
55 – 59
70
60 – 64
60
65 – 69
65
70 – 74
35
75 – 79
30
80 – 84
25
84 – 89
10
> 90
2
nach: Lota AS, Halliday B, Tayal U, et al. Abstract 11463: Epidemiological Trends and Outcomes of Acute Myocarditis in the National Health Service of England. Circulation. 2019;140(Suppl_1):A11463-A11463. doi:10.1161/circ.140.suppl_1.11463
Das sind wie gesagt Daten zu Krankenhausaufnahmen. Zu Prävalenzen findet man wenig, aber z.B. in der Review-Arbeit von Lampejo et al. eine Prävalenzangabe der Myokarditis von 36,5/100.000 bezogen auf die Gesamtbevölkerung Großbritanniens.
Das Ergebnis von Autopsiestudien ist uneinheitlich. Eine Studie zeigte, dass eine Myokarditis bei jedem Dritten Wettkampf-Athleten, der an einem plötzlichen Herztod verstorben ist, vorlag. In anderen Arbeiten zu jungen Männern mit plötzlichem Herztod finden sich Angaben zwischen 2 und 42%, bei denen eine Myokarditis bestand.
Die Prognose der Myokarditis ist abhängig von der Verlaufsform und Schwere der Erkrankung. Je nach Studie wird berichtet, dass bis 25% der Erkrankten eine dauerhafte kardiale Dysfunktion entwickeln. Davon versterben zwischen 12 und 25% oder entwickeln eine terminale Herzinsuffizienz (Kindermann et al.).
Pathophysiologie
Wie es genau zu einer Myokarditis kommt, ist für den Menschen sehr schlecht untersucht. Die meisten Arbeiten beschäftigen sich mit dem Mausmodell der Coxsackie-Virus-Myokarditis, die aber ja in der Realität gar nicht mehr der häufigste Myokarditis-Verursacher ist. Zudem scheinen sich die pathophysiologischen Prozesse auf Zellebene je nach ursächlichem Erreger doch zu unterscheiden.
Beim Coxsackie-Virus scheint es so zu sein: Das Virus wird über einen spezifischen Rezeptor in die Herzmuskelzellen aufgenommen und vermehrt sich dort. Hierunter kommt es zur akuten Myokarditis, die nur wenige Tage andauert. Nun kommt die körpereigene Abwehr ins Spiel, vor allem T-Zellen wandern ins Herzmuskelgewebe ein, diese locken dann mit einer vermehrten Zytokinausschüttung Makrophagen und Monozyten an und später auch B-Zellen, die Antikörper produzieren. Diese Phase nennt man subakute Myokarditis. Kommt es durch autoimmunologische Prozesse zu einer anhaltenden chronischen Entzündungsreaktion, spricht man von einer chronischen postinfektiösen Myokarditis. Diese chronischen Verlaufsformen sind es, die vermutlich in erster Linie zur Kardiomyopathie und Herzinsuffizienz führen.
Symptome einer Myokarditis
Dadurch, dass sehr verschiedene Strukturen des Herzmuskels an einer Myokarditis beteiligt sein können, divergieren die Symptome erheblich. Sie reichen von Herzklopfen (Palpitationen), über Herzrhythmusstörungen bis hin zu Herzinfarkt- und Herzinsuffizienz-Symptomen.
Kinder neigen zu rasch fulminant verlaufenden Myokarditiden, dann aber mit in der Regel guter Erholung im Verlauf trotz zwischenzeitlicher Intensivpflichtigkeit. Bei Erwachsenen scheinen milde Verläufe zu überwiegen, die dafür aber recht häufig übersehen werden.
Diagnosestellung
Neben den oft wenig eindeutigen Symptomen ist das Problem der Diagnosestellung einer Myokarditis der zweite Grund für die anzunehmende Unterdiagnostik.
Als Goldstandard galt ganz lange die Endomyokardbiopsie (EBM), die aber im wahren Leben nur relativ selten durchgeführt wurde. Das ist insofern schade, weil die EBM das einzige Verfahren ist, was durch die Histologie, die gewonnen wird eine genaue, ätiologische Diagnose ermöglicht und hierüber auch die seltenen Formen, z.B. im Rahmen einer TBC oder Sarkoidose detektieren kann.
Laborchemisch ist vor allem der Troponin-Wert für die akute Myokarditis aussagekräftig, insbesondere wenn auch Anamnese und Symptome auf eine Myokarditis hindeuten, die übrigen „Herzenzyme“ aber nicht. Bei der subakuten und chronischen Verlaufsform hilft einem das Troponin hingegen nicht weiter. Die Bestimmung von Virusserologien aus dem Blut wird routinemäßig nicht empfohlen, weil ja viele Myokarditiden erst mit einer gewissen zeitlichen Verzögerung nach der eigentlichen Infektion auftreten und zudem viele der in Frage kommenden Viren endemisch mit einer extrem hohen Durchseuchung und entsprechend positiven Antikörper-Titern sind und so die Serologie oft wenig aussagekräftig bleibt.
Im klinischen Alltag haben die transthorakale Echokardiographie und das Kardio-MRT in den letzten Jahren bei der Fragestellung nach einer Myokarditis einen großen, wenn nicht den größten, Stellenwert bekommen. Insbesondere das Kardio-MRT hat die EBM in vielen Fällen als Haupt-Untersuchungsverfahren abgelöst, so dass die EBM vor allem in „Spezialfällen“, wenn z.B. eine Tuberkulose oder Sarkoidose vermutet wird, angewendet wird.
Nachtrag vom 18.02.2022: Wenn man sich ein bisschen mit Kardio-MRT beschäftigt, taucht dort immer wieder ein Begriff auf: Das late gadolinium enhancement (LGE). Gadolinium ist im MRT-Kontrastmittel enthalten, es geht also um eine „späte“ (spät in der MRT-Messung) Kontrastmittelaufnahme im Herzen. LGE ist mit einem signifikant schlechterem Outcome und dem Auftreten von Herzrhythmusstörungen im Verlauf assoziiert (vgl. Tschöpe et al.).
Das EKG selber hat keine besonders große und insbesondere keine spezifische Aussagekraft, wenn nicht parallel Herzrhythmusstörungen auftreten, die man erkennen und behandeln muss.
In der Summe ist die Diagnosestellung einer Myokarditis nicht trivial. Auch ein auffälliges Kardio-MRT alleine belegt noch lange keine Myokarditis. Somit ist nur die Gesamtbetrachtung sinnvoll: Mit der Betrachtung von Troponin, CRP, Klinik, EF in der Echokardiographie, Kardio-MRT, etwaigen Rhythmusstörungen und dem klinische Verlauf lässt sich einigermaßen zuverlässig die Diagnose stellen.
Behandlung der Myokarditis
Es gibt – wenn kein spezifischer behandelbarer Erreger vorhanden ist – keine kausale Therapie der Myokarditis. Prinzipiell wird körperliche Schonung empfohlen, bei Leistungssportlern für 3-6 Monate. Selbst bei eingeschränkter Pumpfunktion des Herzens kommt es in 60% der Fälle zu einer spontanen und weitestgehenden klinischen Besserung.
Besteht eine Herzinsuffizienz, so wird eine Standard-Herzinsuffizienz-Therapie empfohlen, wobei zu beachten ist, dass die verwendeten Paper noch nicht die neuen Empfehlungen der Herzinsuffizienz-Therapie der neuen ESC-Leitlinie (Link) berücksichtigen.
Kritisch kranke Patienten mit einer Myokarditis müssen auf einer Intensivstation behandelt werden, bei hämodynamischer Instabilität brauchen sie medikamentöse Kreislaufunterstützung und bei schweren Verläufen auch eine ECMO. ECMO ist aber nicht gleich ECMO. Von dieser gibt es wiederum verschiedene „Versionen“ je nach genauer Indikationsstellung, wobei dies an dieser Stelle vermutlich zu weit führt.
Wo man weiterlesen kann
Tschöpe C, Ammirati E, Bozkurt B, et al. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol. 2021;18(3):169-193. doi:10.1038/s41569-020-00435-x
Kindermann I, Ukena C, Mahfoud F, Böhm M, Yilmaz A, Klingel K. Myokarditis-Update. Kardiologe. 2016;10(5):311-330. doi:10.1007/s12181-016-0084-2
Lampejo T, Durkin SM, Bhatt N, Guttmann O. Acute myocarditis: aetiology, diagnosis and management. Clin Med. 2021;21(5):e505-e510. doi:10.7861/clinmed.2021-0121
Myokarditis durch SARS-CoV-2
Vorwort von Lars Fischer
Angesichts der ja doch bescheiden Erkenntnisse, die wir so zur „normalen“ Myokarditis haben, ist es extrem wichtig, sich den weisen Thread von Lars Fischer zu COVID-19-Wissenschafts-Themen vor Augen zu halten, wenn man sich jetzt dem Thema COVID-Myokarditis und danach der Myokarditis nach COVID-Impfung widmet:
Kurzer Hinweis zum Umgang mit beruhigenden und beunruhigenden Forschungsergebnissen aller Art über #COVID19.
In den letzten 2 Jahren wurde darüber gefühlt mehr geforscht als über alle anderen Infektionskrankheiten in den letzten 50. Das hat zwei Konsequenzen: 1/4
Häufiges ist häufig: Epidemiologie der COVID-Myokarditis
Zu dem wenigen, was wir über die Myokarditis wissen gehört, dass in erster Linie junge Männer betroffen sind und Myokarditiden gerne nach Virusinfekten auftreten. Es wäre also hochplausibel, wenn das bei COVID-19 nicht anders wäre.
Studien mit US-Uni-Athleten
Erstaunlich viele Daten (allerdings sind das größtenteils Untersuchungen ohne Kontrollgruppe) gibt es zu Athleten mit COVID-Infektion, vermutlich, weil hier das Thema kardiopulmonale Belastbarkeit eine ganz entscheidende Rolle spielt:
In einer Studie von Daniels et al. mit 1.597 Athleten von US-Universitäten, die eine COVID-Infektion erlitten hatten, wiesen 5 Probanden (0,31%) klinische Zeichen einer Myokarditis auf. 37 (2,3%) der Probanden hatten ein pathologisches Kardio-MRT, welches auf eine Myokarditis hindeutete. 73% der Betroffenen waren männlich. Das heißt, dass ein Großteil der COVID-Myokarditiden in dieser Studie klinisch stumm bzw. sehr symptomarm verlaufen sind, so dass sie ohne Kardio-MRT nie als solche identifiziert worden wären. Ebenfalls 73% Studienteilnehmer erhielten innerhalb von 10 Wochen ein Verlaufs-MRT, bei dem sich bei allen die T2-Läsionen zurückbildeten und bei 40,7% die gesamten MRT-Auffälligkeiten.
In einer anderen Studie mit US-Sportlern (Moulson et al.) wurden von 3.384 SARS-CoV-2 positive Athleten nach einem Screening 2.820 in die Studie eingeschlossen. Zwei Drittel waren auch hier Männer, das mittlere Alter lag bei 20 Jahren. 337 Probanden (13%) berichteten von Zeichen, die für eine Myokarditis sprechen könnten, wie Brustschmerzen, Atemnot, Palpitationen und Belastungsdyspnoe. Bei 21 Studienteilnehmern (0,7%) wurde eine Myokarditis diagnostiziert. Auch hier gelang die Diagnose nicht mit einem einzelnen Test, sondern entweder in der Kombination EKG, Troponin-Bestimmung und Kardio-MRT oder mit zusätzlicher transthorakaler Echokardiographie. Das Kardio-MRT war auch hier das zuverlässigste diagnostische Instrument. Von der gesamten Kohorte hatten 4,5% irgendeine kardiologische Auffälligkeit, 2,7% Auffälligkeiten die initial eine Myokarditis vermuten ließen. Keiner der Myokarditis-verdächtigen Studienteilnehmer entwickelte in einem Zeitraum von 130 Tagen Nachbeobachtungszeit relevante kardiologische Ereignisse, in der Gesamtkohorte der SARS-CoV-2-positiven Probanden gab es einen plötzlichen Herzstillstand, der Proband wurde erfolgreich reanimiert. Ein Kardio-MRT (am 17. Tag nach Infektion) ergab bei diesem Probanden keine Auffälligkeiten. Insgesamt 10 Athleten wurden auf Grund nicht-kardiologischer COVID-Beschwerden im Krankenhaus behandelt, 5 in der ZNA, 5 wurden stationär aufgenommen.
Es gibt tatsächlich noch etliche weitere Studien zu Athleten mit COVID-19 (warum auch immer), zum Beispiel Starekova et al. oder Clark et al.. Auch hier finden sich niedrige einstellige Prozentangaben von Myokarditiden (meist 1-3%) nach COVID-19-Infektion in diesen – meist – jungen Altersgruppen.
Bevölkerungsweite Übersichtsarbeiten
Vor wenigen Tagen wurde in nature medicine eine große Risikoanalyse zu kardiovaskulären COVID-Langzeitfolgen mit Gesundheitsdaten der US Veteranen-Vereinigung veröffentlicht. Untersucht wurde eine Kohorte von 153.760 Veteranen mit COVID-19-Infektion über ein Jahr, welche man mit zwei großen Kontrollgruppen verglich. 5,5 Millionen nicht-COVID-infizierte Veteranen und eine Gruppe von 5,8 Millionen Veteranen aus der Prä-COVID-Ära. Dadurch, dass die ersten 30 Tage nach COVID-Infektion nicht betrachtet wurden, konnte man akute kardiovaskuläre Komplikation ausschließen. Lars Fischer stellt die Studie auch gut verständlich bei spektrum.de vor: Link. Eine kritischere Einordnung der Studie findet sich ansonsten hier: Link. Zum Thema Myokarditis stellen die Autoren ein über 5 x höheres Risiko nach COVID-Infektion fest. Allerdings – und das führt teilweise zur Verwirrung – ist die absolute Fallzahl der Myokarditis immer noch gering. Die „zusätzliche“ Prävalenz (hier als „excess burden“ angegeben) liegt bei 0,31/1.000 Erkrankte.
Eine große retrospektive Studie stammt von Buckley et al. und wurde auch im Eingangs erwähnten Thread von narkosedoc zitiert. Die Autoren verglichen Gesundheitsdaten aus dem TriNetX-Gesundheitsforschungs-Netzwerk (Link) von COVID-Infizierten mit einer Myo- oder Perikarditis mit einer 1:1-gematchten Kohorte COVID-positiver Probanden ohne Myo-/Perikarditis und mit einer Prä-COVID-Kohorte mit einer Lungenentzündung anderer Genese. Von den 718.365 eingeschlossenen Probanden entwickelten 35.820 (5,0%) eine Myokarditis, 10.706 (1,5%) eine eine Perikarditis.
Interessant ist die Altersverteilung der Myokarditis-Betroffenen, die analog zu dem, was wir schon kennen ist: In der Alterskohorte unter 45 Jahren traten 20.774 Myokarditis-Fälle auf, im Alter zwischen 45 und 70 Jahren 14.444 und über 70 Jahren 5.556 Myokarditiden. In dieser Studie waren mehr Frauen als Männer eingeschlossen, das Durchschnittsalter lag bei 47,4 Jahren, also deutlich höher als in den Studien mit Athleten. Wenig überraschend sind die höheren Raten von Komorbiditäten als in den Athleten-Studien: 44% der COVID-Patienten mit Myokarditis litten zusätzlich an einer vorbestehenden Atemwegserkrankung, 37% an einer Erkrankung des Nervensystems, 31% hatten einen Bluthochdruck, 17% einen Diabetes mellitus und immerhin 7,5% eine vorbestehende Herzinsuffizienz. Das Outcome dieser Patienten-Population war dementsprechend schlechter als das der Athleten: Die 6-Monats-Mortalität der Probanden mit Myokarditis betrug 3,9%, die der Kontrollgruppe 2,9%, die zusätzliche Mortalität durch die Myokarditis, die excess mortality dementsprechend 1%. Patienten mit einer Myokarditis erlitten signifikant häufiger Herzinfarkte und Schlaganfälle als die der Kontrollgruppe.
Die Studie unterscheidet zwischen einer Myokarditis und einer Perikarditis. Die Perikarditis trat seltener auf, war aber mit einem schlechteren Outcome und einer insgesamt höheren Mortalität assoziiert.
Weitere Studien
Die Arbeit von Sawalha ist eine Fallsammlung von 14 Patienten, die im Rahmen einer COVID-19-Infektion mit stationärer Behandlungsbedürftigkeit eine Myokarditis entwickelt hatten. Auch hier waren überwiegend Männer betroffen (58%), das mittlere Erkrankungsalter lag bei 50,4 Jahren und damit höher als in den Athleten-Studien. 91% der Probanden hatten eine Troponin-Erhöhung, 50% EKG-Auffälligkeiten. Die insgesamt schwerer betroffenen COVID-Patienten mussten in der Hälfte der Fälle mittels medikamentöser Kreislaufunterstützung auf der Intensivstation behandelt werden, 17% mittels ECMO. Mele et al. haben wiederum eine Übersicht verschiedener Fallsammlungen erstellt, in der die Arbeit von Sawalha et al. wiederum mit erfasst ist. Hier wird noch einmal die Schwierigkeit betont, dass sich viele zunächst Myokarditis-verdächtige klinische Fälle nicht als solche bestätigen lassen.
Hanneman et al. untersuchten 47 COVID-Patienten nuklearmedizinisch mittels PET des Herzens. Die Studienteilnehmer waren eher jung (mittleres Alter 43 Jahre) und leicht betroffen. 85% hatten ihre COVID-Infektion in der Häuslichkeit auskuriert. Bei 17% der untersuchten Probanden ließ sich eine Myokarditis mittels PET feststellen, im Mittel zwei Monate nach der COVID-Infektion. Bei allen Studienteilnehmern besserten sich die auffälligen Befunde innerhalb von 52 Tagen nach Erstuntersuchung oder waren ganz rückläufig.
Wo man weiterlesen kann
Buckley BJR, Harrison SL, Fazio‐Eynullayeva E, Underhill P, Lane DA, Lip GYH. Prevalence and clinical outcomes of myocarditis and pericarditis in 718,365 COVID‐19 patients. Eur J Clin Invest. 2021;51(11). doi:10.1111/eci.13679
Daniels CJ, Rajpal S, Greenshields JT, et al. Prevalence of Clinical and Subclinical Myocarditis in Competitive Athletes With Recent SARS-CoV-2 Infection: Results From the Big Ten COVID-19 Cardiac Registry. JAMA Cardiol. 2021;6(9):1078-1087. doi:10.1001/jamacardio.2021.2065
Moulson N, Petek BJ, Drezner JA, et al. SARS-CoV-2 Cardiac Involvement in Young Competitive Athletes. Circulation. 2021;144(4):256-266. doi:10.1161/CIRCULATIONAHA.121.054824
Xie Y, Xu E, Bowe B, Al-Aly Z. Long-term cardiovascular outcomes of COVID-19. Nat Med. Published online February 7, 2022. doi:10.1038/s41591-022-01689-3
Weitere Literatur (keine Weiterlese-Tips)
Clark DE, Parikh A, Dendy JM, et al. COVID-19 Myocardial Pathology Evaluation in Athletes With Cardiac Magnetic Resonance (COMPETE CMR). Circulation. 2021;143(6):609-612. doi:10.1161/CIRCULATIONAHA.120.052573
Hanneman K, Houbois C, Schoffel A, et al. Combined Cardiac Fluorodeoxyglucose–Positron Emission Tomography/Magnetic Resonance Imaging Assessment of Myocardial Injury in Patients Who Recently Recovered From COVID-19. JAMA Cardiology. Published online January 12, 2022. doi:10.1001/jamacardio.2021.5505
Mele D, Flamigni F, Rapezzi C, Ferrari R. Myocarditis in COVID-19 patients: current problems. Intern Emerg Med. 2021;16(5):1123-1129. doi:10.1007/s11739-021-02635-w
Starekova J, Bluemke DA, Bradham WS, et al. Evaluation for Myocarditis in Competitive Student Athletes Recovering From Coronavirus Disease 2019 With Cardiac Magnetic Resonance Imaging. JAMA Cardiol. 2021;6(8):945. doi:10.1001/jamacardio.2020.7444
Sawalha K, Abozenah M, Kadado AJ, et al. Systematic Review of COVID-19 Related Myocarditis: Insights on Management and Outcome. Cardiovascular Revascularization Medicine. 2021;23:107-113. doi:10.1016/j.carrev.2020.08.028
Myokarditis nach COVID-Impfung
Noch mal ein Vorwort von Lars Fischer
Für keinen Impfstoff den wir je verimpft haben, hatten wir eine derart breite, dauerhafte und intensive öffentliche und wissenschaftliche Diskussion. Zu keinem Impfstoff gibt es so viele Paper, Surveillance und Social Media-Aufmerksamkeit. Das bedeutet, dass ein Underreporting von Impfnebenwirkungen bei COVID-Impfstoffen ziemlich unwahrscheinlich erscheint. Das bedeutet aber auch, dass – auch hier – der direkte Vergleich mit anderen Impfstoffen und ihren Nebenwirkungen nicht so einfach gezogen werden kann und es bedeutet, dass wir noch mal den Lars Fischer-Thread wirken lassen müssen, bevor es weiter geht:
Kurzer Hinweis zum Umgang mit beruhigenden und beunruhigenden Forschungsergebnissen aller Art über #COVID19.
In den letzten 2 Jahren wurde darüber gefühlt mehr geforscht als über alle anderen Infektionskrankheiten in den letzten 50. Das hat zwei Konsequenzen: 1/4
Myokarditien – so haben wir ja schon erfahren – werden durchaus als Impfnebenwirkung berichtet. Nach allem, was bislang in diesem Artikel so stand wäre eine Sache ziemlich plausibel: Junge Männer müssten besonders häufig betroffen sein. Und wie wir alle wissen, ist es auch so. Wenn wir jetzt einen Vergleich ziehen wollen und alle Underreporting– und Overreporting-Probleme außer Acht lassen, brauchen wir irgendwoher Daten. Da gibt es schlussendlich zwei Möglichkeiten: Paper und Arzneimittelsicherheits-Überwachung.
Daten vom Paul-Ehrlich-Instituts
Die in Deutschland gemeldeten Verdachtsfälle von Impfnebenwirkungen landen beim Paul-Ehrlich-Institut (PEI), dass regelmäßig Sicherheitsberichte herausgibt. Der derzeit aktuellste stammt vom 07.02.2022 (Link) und beinhaltet eingegangene Meldungen über Impfkomplikationen bis zum 31.12.2021.
Das PEI schlüsselt die Häufigkeiten einer Myokarditis in dieser Tabelle nach den beiden mRNA-Impfstoffen von BioNTech und Moderna auf:
Leider wird nirgendwo ersichtlich, wie hoch die Zahl der verimpften Impfdosen je Altersgruppe ist, so dass selber man keine Prävalenz für alle Altersgruppen ausrechnen kann. Das PEI gibt nur eine Prävalenz nach Booster von 0,38 pro 100.000 Impfungen für BioNTech und 0,34 für Moderna an. Für die Gruppe der jungen Männer zwischen 18 und 29 Jahren wird sie mit 1,11 pro 1.000.000 Impfungen für BioNTech und mit 2,98 für Moderna angegeben.
Insgesamt wird aber deutlich, dass in erster Linie junge Männer nach der zweiten Impfung betroffen zu sein scheinen. Die meisten Fälle scheinen an Tag 1-4 nach Impfung aufzutreten, dann fällt die Zahl der berichteten Myokarditiden stark ab. Je sieben Myokarditis-assoziierte Todesfälle wurden in Deutschland für den BioNTech und den Moderna-Impfstoff gemeldet. Bei drei Fällen nach BioNTech-Impfung und keinem nach Moderna-Impfung wurde eine Assoziation seitens des PEI als möglich erachtet.
Bei Jugendlichen zwischen 12 und 17 Jahren wurden in Deutschland bislang 147 Verdachtsfälle, ganz überwiegend bei Jungen und nach der zweiten Impfung, gemeldet. Die Melderate für Myokarditiden betrug bei männlichen Jugendlichen 5,1 pro 100.000 Impfungen über beide Impfdosen und 2,0 nach der ersten und 8,6 nach der zweiten Dosis. Für weibliche Jungendliche lang sie stabil bei 0,6 pro 100.000 Impfungen.
Bei Kindern unter 12 Jahren lagen bis zum 31.12. (wen wundert es bei einem Impfstart wenige Tage zuvor) keine Daten zu Myokarditiden vor.
Die britische Arzneimittel-Surveillance
Aktueller und ähnlich umfassend wird man – auch bei diesem Thema – von den britischen Gesundheitsbehörden informiert. Die Medicines and Healthcare products Regulatory Agency (MHRA) veröffentlicht Sicherheitsberichte, wie wir sie auch vom PEI kennen. Hier findet man den aktuellen, für diesen Beitrag verwendeten: Link. Bis zum 02.02.2022 sind dort 1.165 Verdachtsfälle von postvaccinalen Myokarditiden (697) und Perikarditiden (468) nach Impfung mit dem BioNTech-Impfstoff eingegangen. Für den AstraZeneca-Impfstoff waren es 214 Myokarditis-Verdachtsfälle und 212 Perikarditisverdachtsfälle und für Moderna 194 Myokarditis- und 111 Perikarditis-Verdachtsfälle. Jeweils vier tödliche Verläufe wurden für den BioNTech- und den AstraZeneca-Impfstoff gemeldet, keine für den Moderna-Impfstoff. Bei den „meisten“ tödlichen Verläufen sei die Grunderkrankung des jeweiligen Impflings ursächlich gewesen, schreibt die MHRA.
Auf die Gesamtpopulation aller Briten über alle Altersgruppen und alle Impfdosen sind das laut MHRA 9 Myokarditiden pro 1.000.000 Impfungen bei BioNTech, 17 Myokarditiden pro 1.000.000 Impfungen bei Moderna und 4 pro 1.000.000 bei AstraZeneca. Die Aufschlüsselung nach Altersgruppen ergibt den von den infektiös bedingten Fällen bekannten Altersgipfel bei jungen Erwachsenen für den BioNTech- und Moderna-Impfstoff, nicht aber für den AstraZeneca-Impfstoff.
Two large European epidemiological studies have estimated the excess risk of myocarditis following vaccination with COVID-19 Pfizer/BioNTech Vaccine and COVID-19 Vaccine Moderna. One study showed that in a period of 7 days after the second dose of COVID-19 Pfizer/BioNTech Vaccine there were about 27 (95% CI 26 – 28) extra cases of myocarditis in 12-29 year old males per million compared to unvaccinated individuals, and for COVID-19 vaccine Moderna there were about 132 (95% CI 130 – 133) extra cases of myocarditis in 12-29 year old males per million. In another study, in a period of 28 days after the second dose of the COVID-19 Pfizer/BioNTech Vaccine there were 57 (95% CI 39 – 75) extra cases of myocarditis in 16-24 year old males per million compared to unvaccinated persons, and for COVID-19 vaccine Moderna. there were 190 (95% CI 96 – 280) extra cases of myocarditis in 16-24 year old males per million individuals compared to unvaccinated individuals. These studies have shown that these events are very rare post vaccination with the mRNA vaccines, and that these events are more frequent in younger males. The findings of these studies are consistent with the trends seen in the Yellow Card data.
Was die MHRA aber auch schreibt ist dies:
It is important to note that Yellow Card data cannot be used to compare the safety profile of COVID-19 vaccines as many factors can influence ADR reporting.
Okay, schauen wir also noch mal in die medizinische Literatur.
Paper
Arzneimittelüberwachung in den USA
In Impfgegner- und -skeptiker-Kreisen wird in den letzten Tagen immer wieder dieser Artikel zitiert, der auf diese Studie von Oster et al. verweist: Link. Auch narkosedoc verwendet dieses Paper in seinem Thread. Die Daten für dieses Paper stammen aus der Arzneimittelüberwachung der USA. 192.405.448 Personen wurden mit 354.100.845 Dosen mRBA-Impfstoff geimpft. Von 1991 Berichten über eine impfassoziierte Myokarditis konnten 1626 verifiziert werden. Auch hier betrafen die Myokarditiden in erster Linie junge Männer. Männer waren auch in dieser Studie ganz überwiegend betroffen (82% der Fälle), das mittlere Alter für eine Myokarditis nach Impfung lag bei 21 Jahren, die Myokarditiden traten innerhalb von 7 Tagen und besonders häufig nach der zweiten Impfung auf. Die Myokarditidis-Raten lagen bei Jungen im Alter von 12-15 Jahren bei 70,7 pro 1.000.000 Impfungen nach der zweiten Impfung und bei 16-17-Jährigen bei 105,9.
BioNTech
Moderna
D1
D2
D1
D2
expected
Männer
Alter
12 – 15
7,06
70,73
/
/
0,53
16 – 17
7,26
105,86
/
/
1,34
18 – 24
3,82
52,43
10,73
56,31
1,76
25 – 29
1,74
17,28
4,88
24,18
1,45
30 – 39
0,54
7,1
3,0
7,93
0,63
40 – 49
0,55
3,5
0,59
4,27
0,78
50 – 64
0,42
0,68
0,62
0,85
0,77
> 65
0,19
0,32
0,18
0,51
Frauen
Alter
12 – 15
0,49
6,35
/
/
0,17
16 – 17
0,84
10,98
/
/
0,42
18 – 24
0,18
4,12
0,96
6,87
0,38
25 – 29
0,26
2,23
0,41
8,22
0,48
30 – 39
0,72
1,02
0,74
0,68
0,47
40 – 49
0,24
1,73
0,18
1,89
0,89
50 – 64
0,37
0,51
0,65
0,43
1,0
> 65
0,08
0,35
/
0,26
/
Angaben per 1 Million Impfdosen. Nach: Oster ME, Shay DK, Su JR, et al. Myocarditis Cases Reported After mRNA-Based COVID-19 Vaccination in the US From December 2020 to August 2021. JAMA. 2022;327(4):331-340. doi:10.1001/jama.2021.24110
Für 676 Impfstoff-assoziierte Myokarditiden lagen klinische Behandlungsdaten vor. Berichtete Symptome waren in erster Linie Brustschmerzen, verminderter Belastbarkeit und Luftnot. Bei nahezu allen Betroffenen waren das Troponin erhöht, in 72% der Fälle das Kardio-MRT auffällig. Die meisten Betroffenen erhielten eine symptomatische Therapie mit NSAR, seltener mit Steroiden oder Immunglobulinen. 12 Betroffene mussten mittels medikamentöser Kreislaufunterstützung behandelt werden, 2 künstlich beatmet werden. Zum Ende der Nachverfolgung durch die Studie waren 98% der Betroffenen aus dem Krankenhaus entlassen und 97% symptomfrei.
Der Impfungs-Infektions-Vergleich
Interessant ist noch die Studie von Patone et al. Sie zeigt zunächst die Dinge, die wir nun schon wissen: In erster Linie sind junge Männer betroffen, vor allem nach der zweiten Impfdosis mit einem mRNA-Impfstoff. Die Autoren vergleichen aber die Rate von Myokarditiden nach COVID-Infektion mit der nach COVID-Impfung und können für die Gesamtpopulation deutlich häufigere Myokarditiden durch die Infektion als durch die Impfung zeigen, nicht aber, wenn man die Gruppe der unter 40-jährigen betrachtet.
aus Patone M, Mei XW, Handunnetthi L, et al. Risks of myocarditis, pericarditis, and cardiac arrhythmias associated with COVID-19 vaccination or SARS-CoV-2 infection. Nature Medicine. Published online December 14, 2021. doi:10.1038/s41591-021-01630-0
Myokarditis-Verlauf nach COVID-Impfung bei Kindern und Jugendlichen
Die Arbeit von Jain et al. beleuchtet den Verlauf impfassoziierter Myokarditiden bei Jugendlichen am Beispiel von 63 Fällen aus den USA. 92% betragen Jungen, das mittlere Alter lag bei 15,6 Jahren. Bei 88% der vermuteten Myokarditiden waren die diagnostischen Kriterien auch erfüllt. Alle Verläufe waren leicht, kein Patient musste intensivmedizinisch behandelt werden. Bei 86% der Betroffenen waren Symptome und auffällige Befunde innerhalb von 35 Tagen komplett regredient.
Dionne et al. haben hier eine kleinere Serie von 15 Kindern veröffentlicht, die im Endeffekt die selben, milden Verläufe zeigte.
Problematisches Preprint: Der Infektions-Impfungs-Myokarditis-Vergleich
Dieses Preprint (Link ) scheint dem Titel nach (Risk of Myocarditis from COVID-19 Infection in People Under Age 20: A Population-Based Analysis) und dem Abstract nach
Results: For the 12-17-year-old male cohort, 6/6,846 (0.09%) patients developed myocarditis overall, with an adjusted rate per million of 876 cases (Wilson score interval 402 – 1,911). For the 12-15 and 16-19 male age groups, the adjusted rates per million were 601 (257 – 1,406) and 561 (240 – 1,313).
For 12-17-year-old females, there were 3 (0.04%) cases of myocarditis of 7,361 patients. The adjusted rate was 213 (73 – 627) per million cases. For the 12-15- and 16-19-year-old female cohorts the adjusted rates per million cases were 235 (64 – 857) and 708 (359 – 1,397). The outcomes occurred either within 5 days (40.0%) or from 19-82 days (60.0%).
Conclusions: Myocarditis (or pericarditis or myopericarditis) from primary COVID19 infection occurred at a rate as high as 450 per million in young males. Young males infected with the virus are up 6 times more likely to develop myocarditis as those who have received the vaccine.
auf den ersten Blick alle offenen Fragen zu beantworten. Aber es gibt Punkte, die exemplarisch zeigen, wann man bei Preprints stutzig werden sollte: Das Preprint wurde vor 9 Monaten zur Begutachtung eingestellt und wurde seither nicht offiziell veröffentlicht. Das verwundert bei der Brisanz und Dringlichkeit der Thematik dann doch. Ein Blick in die Kommentare unter dem Preprint enthüllt dann aber auch warum: Es scheint ein massives Problem bei der Statistik der Fallzahlenberechnung zu geben.
Fazit: Nicht verwenden, nicht zitieren, abwarten, ob das Paper durch die Autoren noch gerettet werden kann.
Wo man weiterlesen kann
Jain SS, Steele JM, Fonseca B, et al. COVID-19 Vaccination–Associated Myocarditis in Adolescents. Pediatrics. 2021;148(5):e2021053427. doi:10.1542/peds.2021-053427
Oster ME, Shay DK, Su JR, et al. Myocarditis Cases Reported After mRNA-Based COVID-19 Vaccination in the US From December 2020 to August 2021. JAMA. 2022;327(4):331-340. doi:10.1001/jama.2021.24110
Patone M, Mei XW, Handunnetthi L, et al. Risks of myocarditis, pericarditis, and cardiac arrhythmias associated with COVID-19 vaccination or SARS-CoV-2 infection. Nature Medicine. Published online December 14, 2021. doi:10.1038/s41591-021-01630-0
Ein Fazit
Das, was alle gerne hätten, das gibt es nicht: Eine Tabelle, in der schön nach Geschlecht und Altersgruppe nebeneinander statistisch belastbare Daten zur „normalen“ Virus-Myokarditis, zur COVID-Myokarditis und zur Impf-Myokarditis aufgeschlüsselt sind.
Nachtrag vom 18.02.2022: Die Paper von Fronza et al. versucht sich mit jeweils sehr kleinen Fallzahlen (21 Probanden mit Myokarditis nach COVID-Impfung, 10 Probanden mit Myoarditis nach COVID-Infektion, 61 mit Myoarditis aus anderen Gründen) an diesem Vergleich (Dank an Jesse Ventura für das Paper). Die Myokarditiden nach Impfung waren im Vergleich zu den COVID-Infektions-Myokarditiden und den „anderen“ Myokarditiden milder, mit weniger Einschränkungen der Pumpfunktion und weniger Auffälligkeiten in der Kardio-MRT-Untersuchung. Allerdings waren sie auch im Schnitt 20 Jahre jünger (Altersdurchschnitt 31 Jahre bei der Impf-Myokarditis, 51-Jahre bei der COVID-Myokarditis, 44 Jahre bei den „anderen“ Myokarditiden. Von den Probanden mit Impf-Myokarditis hatten 81% ein LGE (siehe oben) und 29% eine eingeschränkte Pumpfunktion. Zur Einordnung: Ein LGE hatten 90% der COVID-Myokarditis-Probanden und 97% der mit einer „anderen“ Myokarditis, eine eingeschränkte Pumpfunktion 50% der Probanden nach COVID und 77% der „anderen“. In einer follow up-Untersuchung waren nach durchschnittlich 22 Tagen die Impf-Myokarditis-Probanden wieder beschwerdefrei. Die Troponin-Werte hatten sich bei 38% normalisiert, bei 43% verbessert. Alle Probanden hatten wieder eine normale Pumpfunktion. Bei keinem Probanden trat im Nachbeobachtungszeitraum eine behandlungsbedürftige Herzrhythmusstörung oder ein andere akute kardiologische Erkrankung auf. Bei den COVID-Probanden und den „anderen“ war der Nachbeobachtungszeitraum jeweils 10 x so lang (211 und 195 Tage, was aber lt. Autoren an der kurzen Verfügbarkeit der COVID-Impfstoffe liegt). Hier traten in beiden Gruppen behandlungsbedürftige kardiologische Erkrankungen auf (bei 30% der COVID-Gruppe und 8% der „anderen“ Myokarditiden). Keiner der Probanden (egal welcher Kohorte) ist im Beobachtungszeitraum verstorben.
Was kann man nun aus den vorgestellten Arbeiten lernen: Männliche Jugendliche und junge Männer haben ein erhöhtes Risiko einer mRNA-COVID-19-Impfstoff-assoziierten Myokarditis, insbesondere nach der zweiten mRNA-Imfpung. Dieses Risiko übersteigt allem Anschein nach das Risiko einer Myokarditis durch COVID-19 selber. Diese Impfstoff-Myokarditiden verlaufen in der Regel mild und heilen gut ab. Für alle anderen Altersgruppen bei Männern gilt das nicht. Hier ist das COVID-Myokarditis-Risiko höher als dass der Impfung. Bei Frauen kann man einen ähnlichen, wenn aber nicht so ausgeprägten Effekt sehen. Wichtig ist auch zu beachten, dass Männer (auch jüngere) statistisch sehr viel schwerere COVID-Verläufe als Frauen haben. Dementsprechend sollten sich auch junge Männer impfen lassen.
Niemand lässt sich gegen COVID-Impfen, weil er isoliert Sorge vor einer COVID-assoziierten Myokarditis hat. Alle lassen sich impfen, damit sie möglichst keinen schweren Verlauf, kein Long Covid und bei Kindern und Jugendlichen kein PIMS bekommen. Seit der Ausbreitung der Delta-Variante gilt ja der berühmte Jens Spahn-Spruch, der in seinem Pathos etwas übertrieben scheint, aber einen wahren Kern hat:
Die Frage ist nicht, bekommt man einfach kein COVID und kann sich wegducken, sondern bekommt man COVID oder ist man geimpft? Auf das Myokarditis-Thema runter gebrochen: Welche Myokarditis-Version hätte man lieber im Fall der Fälle? Mit Omikron ist selbst diese brachiale Weisheit mehr oder weniger hinfällig. Sehr viele Menschen werden auch geimpft COVID bekommen.
Dennoch erscheint es sinnvoll, dass es Überlegungen gibt, den Impfabstand zumindest bei jungen Männern zu verlängern und auch die Dosis ggfs. zu reduzieren. Hilfreich hierfür ist sicherlich die COV-BOOST-Studie von Munro et al., die entsprechende Signale (bei Probanden > 30 Jahren) zeigen konnte oder auch dieses Preprint (Link) von Buchan et al. (Danke an Dr.Rup für das Paper). In Großbritannien wurde aus genau diesen Überlegungen ein Impfabstand von 12 Wochen bei der COVID-Impfung von 5-11-Jährigen empfohlen (Link).
Wo man weiterlesen kann
Buchan SA, Seo CY, Johnson C, et al. Epidemiology of myocarditis and pericarditis following mRNA vaccines in Ontario, Canada: by vaccine product, schedule and interval. medRxiv. Published online January 1, 2021:2021.12.02.21267156. doi:10.1101/2021.12.02.21267156
Fronza M, Thavendiranathan P, Chan V, et al. Myocardial Injury Pattern at MRI in COVID-19 Vaccine–associated Myocarditis. Radiology. Published online February 15, 2022:212559. doi:10.1148/radiol.212559
Munro APS, Janani L, Cornelius V, et al. Safety and immunogenicity of seven COVID-19 vaccines as a third dose (booster) following two doses of ChAdOx1 nCov-19 or BNT162b2 in the UK (COV-BOOST): a blinded, multicentre, randomised, controlled, phase 2 trial. The Lancet. 2021;398(10318):2258-2276. doi:10.1016/S0140-6736(21)02717-3
Marc Hanefeld, Allgemeinmediziner aus Bremervörde, der bei Twitter unter dem Namen @Flying__Doc auftritt, hat heute folgenden Thread zum Thema No Covid verfasst:
Habe Bekannte im Gastronomie-Sektor. #2GPlus bedeutet für die, dass Einnahmeverluste drohen, ohne Kompensation.
#NoCovid damals (von Vielen unverstanden!) beinhaltete auch zeitlich begrenzten Lockdown: Recht auf finanzielle Kompensation, frühere Normalisierung.
Es wird deutlich, dass die #NoCovid-Hasser, die immer sagten „Aber die Wirtschaft!“ nun vor einer Situation stehen, in der die Kleinunternehmer in gewissen Branchen in die Knie gehen.
Das zeigt auch ganz deutlich die Moral dieser Bubble. Die ist auf Großwirtschaft zentriert.
Nicht falsch verstehen: #NoCovid (besser: #LowCovid) ist jetzt aktuell total unrealistisch, gerade angesichts der medialen und politischen Kommunikation. Ich fordere das zum jetzigen Zeitpunkt nicht. Aber ich darf schon mal sagen: Wie dumm kann eigentlich eine Gesellschaft sein?
Ob aus dem FDP- oder aus dem einfachen Rechtsrad.-Umfeld: Die Leute, die ein aktives Pandemie-Management ablehnen und bekämpfen, verursachen genau damit weit mehr Schaden. Wirtschaftlich und medizinisch. Auch Tote. Wer dem das Wort redet, verhält sich gesellschaftsschädigend.
Sicherheitshalber: Wer das hier nutzt für hirnloses NoCovid-Bashing, der wird ohne Vorwarnung geblockt. Ich halte meine Timeline sauber.
Ich versuche mich mal an einer persönlichen Erwiderung und ich hoffe wir bekommen das höflich und ohne gegenseitiges beschimpfen und geblocke hin. Ich gehöre zu denen, die das No Covid-Konzept ablehnen und nicht, weil ich Großkonzernen oder einem Raubtierkapitalismus den Mund rede, sondern weil ich es für ein was Freiheitsrechte betrifft höchst bedenkliches und gefährliches Konzept halte. Und damit für „gesellschaftsschädigend“.
Und ja, wir können das ganze abkürzen, mein Freiheitsverständnis ist offenbar primitiv und nur die FDP und Lars Weisbrod teilen das, usw. Wenn man es schafft, sich nicht an die ins lächerliche gehende Dichotomie zwischen Querschwurblern und Rotknödel zu verlieren, könnte man aber sogar ins Gespräch kommen.
Also, No Covid kann man ja unter 2 Gesichtspunkten vertreten:
Als Übergangsstrategie bis wirksame Impfstoffe zur Verfügung stehen
Als dauerhaftes Pandemie-Eindämmungs-Konzept
Viele Menschen, die ich kenne, auch und insbesondere Mediziner, haben No Covid entsprechend Punkt 1 vertreten. Deutlich weniger verstehen es (außerhalb von Corona-Twitter) entsprechend Punkt 2.
SARS-CoV-2 is here to stay
Ich denke, NoCovid ist nicht auf Grund der „medialen und politischen Kommunikation“ unrealistisch, sondern in erster Linie dadurch, dass SARS-CoV-2 unter allgemein wissenschaftlich akzeptierten Annahmen als weltweit pandemisch gewordenes Virus nicht mehr zu eliminieren / eradizieren ist. Bis zu welchem Zeitpunkt dies realistisch gewesen ist, darüber scheiden sich die Geister, ich persönlich halte die Einschätzung von Francois Balloux für recht überzeugend:
I appreciate covid elimination / eradication was a tenable position until late summer 2020. I'm just intrigued that anyone believes it may still be now, and even more so, that some feel the emergence of yet another possibly more transmissible lineage supports those views. 3/
Auch von Anfang an wäre die SARS-CoV-2-Elimination ein ambitioniertes Ziel gewesen, insbesondere wenn SARS-CoV-2 einem tierischen Wirt entsprungen ist und die Laborhypothese nicht stimmt und es so ein dauerhaftes „Nachschub-Reservoir“ gibt. Seit überzeugende Hinweise bestehend, dass SARS-CoV-2 bei Omikron vom Menschen auf einen tierischen Zwischenwirt und zurück zum Menschen gesprungen ist (vgl. Wei, C., Shan, K.-J., Wang, W., Zhang, S., Huan, Q., & Qian, W. (2021). Evidence for a mouse origin of the SARS-CoV-2 Omicron variant. Journal of Genetics and Genomics, xxxx. https://doi.org/10.1016/j.jgg.2021.12.003) ist die Elimination des Virus wohl kaum mehr zu schaffen. Vor allem, wenn man bedenkt, dass SARS-CoV-2 auch in den Ländern heimisch geworden sind, wo es weder politisch, wirtschaftlich und medizinisch die Strukturen gibt, das Virus aktiv und effektiv zu bekämpfen.
Das No Covid-Konzept
Das Konzept von No Covid (Link) beruht im Wesentlichen auf dem Gedanken, relativ kleinräumig die Zahl der Neuinfektionen mit SARS-CoV-2 nahezu komplett auf null zu senken, bzw. keine nicht nachvollziehbaren Infektionsketten mehr zu haben und dann diese COVID-freien Zonen nach und nach zu größeren zusammenzufügen. Bei neuen Infektionen unklaren Ursprungs erfolgen dann umgehend harte Eindämmungsmaßnahmen um die Infektionsketten wieder zu durchbrechen. Man verspricht sich dadurch eine insgesamt kürzere Zeit mit Einschränkungen, zwischen den Lockdowns ein weitestgehend normales Leben und sowohl was die Zahl an COVID-Toten, als auch was die wirtschaftlichen Auswirkungen betrifft ein besseres Outcome, als wenn man die Infektionen „einfach laufen lässt“. Um das zu erreichen, gibt es zwei Hauptstrategien (Link):
Grüne und rote Zonen und
Test Trace and Isolate (TTI)
Grüne und rote Zonen
Die kleinräumigen Gebiete, in denen durch nicht-pharmazeutische Interventionen (NPI) (Lockdown, Abstandsgebot, Masken, Kontaktbeschränkungen usw.) eine Null-Inzidenz erreicht wurde, werden grüne Zonen genannt. Diese werden gegenüber Vireneintragungen von außen relativ hermetisch abgeriegelt, d.h. bis auf unbedingt notwendigen Transit-, Liefer- und Pendelverkehr (der mit Test- und ggfs. Quarantänemaßnahmen belegt wird) wird die Mobilität in rote Zonen, wo eben höhere Inzidenzen vorherrschen unterbunden. Sie ist im wesentlichen nur zwischen grünen Zonen gestattet.
Als Vorbild werden in der Regel Australien und Neuseeland genannt und z.B. hier der Maßnahmenplan von Melbourne als Vorbild für einen effektiven Lockdown genannt, der bis zu einer 7-Tageinzidenz von weniger als 10/100.000 das Verlassen des eigenen Hauses nur aus triftigem Grund gestattet und Besuche nur bei Alleinlebenden (1 Person), geschlossene Schulen usw. Erst bei einer 7-Tageinzidenz von null über vier Wochen wird „schrittweise“ auch das Arbeitsleben als letzter Schritt normalisiert.
Test Trace and Isolate
Damit ist die Kontaktnachverfolgung mit Quarantänisierung von Kontaktpersonen und Isolation von Infizierten gemeint. Damit eine Nullinzidenz aufrechterhalten werden kann, muss diese sehr effektiv und auch sehr proaktiv stattfinden, im Zweifelsfall werden eher zu viel als zu wenig Quarantänemaßnahmen verhängt. Das No Covid-Testkonzept findet man hier.
No Covid und Freiheitsrechte
Um das No Covid-Konzept aufrechtzuerhalten, müssen Grundrechte eingeschränkt werden, insbesondere die Freizügigkeit. Das fängt bei der Mobilität an, geht weiter bei Dingen wie Ausgangssperren und endet bei Kontaktbeschränkungen. Dazu kommt eine umfassende staatliche Kontrollmöglichkeit darüber, mit wem ich mich wann getroffen habe (denn die brauche ich für TTI). Und jetzt kommen wir zurück auf die Szenarien 1 und 2 von weiter oben.
Szenario 1, dass der zeitlich begrenzten Umsetzung bis zur Verfügbarkeit und Grundimmunisierung weiter Teile der Bevölkerung und
Szenario 2, dass der dauerhaften Pandemie-Kontrolle durch No Covid.
Schon bei Szenario 1 muss sich für mich sagen, empfinden ich den staatlichen Eingriff als übergriffig und totalitär. Jetzt komme ich aus einer politischen Ecke, in der die Einrichtung der Gefahrengebiete auf St. Pauli und im Schanzenviertel Anfang 2014 als Reaktion auf mehrere Rote-Flora- und Lampedusa-Flüchtlings-Demonstrationen (und ich meine auch Esso-Häuser-Anti-Gentrifizierungs-Sachen waren auch dabei) als unzumutbarer staatlicher Übergriff wahrgenommen wurde, ebenso die Polizeimaßnahmen um den G20-Gipfel drei Jahre später. Und selbst wenn man – wie ich – mit vielen kapitalismuskritischen (oder noch schlimmer antiimperialistischen) Demonstrationen nichts anfangen konnte, war klar, dass es ein Unding ist eine Stadt in einen Ausnahmezustand zu versetzen und Transferrouten für das who is who der Despoten, Populisten und Egomanen dieser Welt freizuprügeln.
Die staatlichen Eingriffe für die No Covid-Strategie betreffen aber nicht nur Hamburg oder Teile davon, sondern das ganze Land, ggfs. die ganze EU. Und sie betreffen nicht nur ein paar Zecken und Journalisten, sondern alle Menschen.
Ich kann – sehr widerstrebend – nachvollziehen, dass der Eingriff in Freiheitsrechte in einer sonst unkontrollierbaren Pandemie von Nöten ist, bis bessere Containment-Strategien zur Verfügung stehen. Von daher kann ich Szenario 1 noch irgendwie akzeptieren, auch wenn ich es persönlich nicht für richtig halte.
Szenario 2 hingegen nicht. Wir haben jetzt hochwirksame Impfstoffe, die insbesondere vulnerable Bevölkerungsgruppen vor schweren COVID-Verläufen mit sehr hoher Wahrscheinlichkeit schützen können. Ja, wir haben eine – im Vergleich zu anderen Ländern mit ähnlicher wirtschaftlicher Leistungsfähigkeit – sehr hohe Rate an ungeimpften Menschen. Trotzdem, lehne ich es ab, meine Freizügigkeit auf Dauer aufzugeben, weil Leute sich – aus aus meiner Sicht irrationalen Gründen – gegen eine Impfung gegen COVID-19 entscheiden. Weil das das Problem dieser Leute ist. Und wenn – nach der aktuellen Welle – es nicht mehr zu befürchten steht, dass das Gesundheitssystem angesichts der Anzahl an kritisch kranken Menschen mit COVID-19 kapituliert, akzeptiere ich diese Freiheitseinschränkungen nicht weiter. Einfach weil es Freiheitseinschränkungen sind und nicht weil ich ein „aktives Pandemie-Management“ ablehne oder ähnliches. Szenario 2 hat aus den genannten Gründen kein absehbares Ende, da SARS-CoV-2 nicht verschwinden wird und es so immer wieder Krankheitswellen und Eintragungen des Virus geben wird. No Covid wäre damit der Eintritt in einen Überwachungsstaat (wenn auch aus „noblen“ Gründen, wie unser jetziger Gesundheitsminister mal bei Twitter geschrieben hat).
Off Topic: Einfluss der No Covid-Protagonisten auf Pandemie-Politik in Deutschland
Es lohnt sich durchaus hier nachzuschauen, welche Wissenschaftler zum No Covid-Kernteam gehören und wer davon vor allem zum Beraterstab der Regierung Merkel gehörte. Aber auch im aktuellen Expertenrat der Bundesregierung (Link) finden sich einige Mitglieder wieder. Es lohnt sich durchaus auch nachzuschauen, welche Test- und Eindämmungskonzepte bei No Covid beschrieben wurden und welche wir aus der Vergangenheit aus praktischer Anwendung kennen. Wenn es heißt, Deutschland habe mit die strengsten und rigidesten Pandemie-Eindämmungsmaßnahmen, gibt es so durchaus eine Idee, warum das so sein könnte.
Vielleicht ist es auch nur Psychohygiene, weil ich eigentlich diese Faktenchecker, Faktenfüchse, Volksverpetzer usw. ganz furchtbar finde, weil da meistens mehr Agenda als Faktencheck hintersteckt. Aber wenn wer Freude dran hat:
22.03.2021
(1) Grosse internationale Studie UK/US hat 6 Monate #LongCovid untersucht. Bisher beste Langzeitstudie dazu. Es fällt auf, dass diejenigen, die Denkstörungen und Merkprobleme nach 1 Mon hatten diese in 6 Monaten nicht losgeworden sind. Was heisst das? https://t.co/vIycT8VBibpic.twitter.com/WMh69BIx27
— Prof. Karl Lauterbach (@Karl_Lauterbach) March 22, 2021
Blog-Beitrag zu dem Thema (Link), bis zu „Oft zitiert und oft kritisiert: Die Studie mit dem Internetfragebogen“ scrollen.
08.05.2021
Die Grafik ist wertvoll und beschreibt die Studienlage. Quintessenz: AstraZeneca lohnt für fast alle mit erhöhtem Infektionsrisiko und für ALLE Älteren. Da Eltern erhöhtes Infektionsrisiko haben lohnt sich schneller AstraZeneca Termin auch für sie. https://t.co/LOH8qyxZwr
— Prof. Karl Lauterbach (@Karl_Lauterbach) May 7, 2021
Blog-Beitrag zu dem Thema, das Thema VITT bei jungen mit AstraZeneca-Geimpften war da schon längst bekannt: Link.
12.05.2021
(1) Diese wichtige Studie aus Kalifornien belegt, was lange vermutet wurde. Covid ist viel mehr eine Gefässkrankheit als eine Lungenkrankheit. Das erklärt auch die Schäden an Nieren, Gehirn und Herz. Das Spike Protein selbst beschädigt die Gefässe. https://t.co/H71qSaul13
— Prof. Karl Lauterbach (@Karl_Lauterbach) May 12, 2021
Nein, keine dieser Schlussfolgerungen kann man nach Lektüre dieses Beitrages https://t.co/HqI3x1nWVs ernsthaft behaupten. Es handelt sich um einen interessanten Beitrag aus der Grundlagenforschung zu #COVID19, in dem ein Pseudovirus in einem Tiermodell (Hamster) verwendet wurde.
Wichtige Studie aus Cambridge von @GuptaR_lab zu Varianten und BionTech. Nach der ersten Impfung wirkt BionTech bei Ü80 Geimpften schlecht. Nach 2er Impfung ist Wirkung da, aber Dauer unklar. Auch Wirkung gegen Delta Variante unklar. Der Herbst ist riskant https://t.co/J9i45dhgxy
— Prof. Karl Lauterbach (@Karl_Lauterbach) June 30, 2021
Schon erstaunlich, wie unterschiedlich man die selbe Studie lesen kann. Ich wäre da eher bei @CarstenWatzl. Und ich finde es beeindruckend, wie man immer wieder Analogien ziehen kann, die das zitierte Material gar nicht hergibt („Der Herbst ist riskant“). #COVID19#Delta#impfenpic.twitter.com/laSdxrBQ8a
Ausführliche Zusammenfassung @GuptaR_lab Cambridge UK weshalb Delta Variante so gefährlich auch für Geimpfte ist. Dazu kommt schwererer Verlauf und schlechte Behandelbarkeit. Die Bevölkerung diesem Virus auszusetzen, wie in UK ab morgen der Fall, ist voll verantwortungslos https://t.co/y1EYeAL6wt
— Prof. Karl Lauterbach (@Karl_Lauterbach) July 18, 2021
1/ Selbst im Abstract steht: „Whilst severe disease in fully vaccinated HCW was rare“. Im Paper auch nichts anderes. Wie kommen Sie darauf, dass der Artikel belegt, dass #Deltavariante auch für #Geimpfte gefährlich sei? #COVID19#Impfen#coronaHH
In UK steigen Infektionszahlen, Krankenhauseinweisungen und Todesfälle. Mehrheit der neuen Krankenhausfälle sind geimpft. Das Risiko, jetzt weiter zu öffnen, wäre mir viel zu gross. https://t.co/hiU4pF33C6
— Prof. Karl Lauterbach (@Karl_Lauterbach) July 19, 2021
Weiterhin ist zu beachten: Wenn 95 % einer Bevölkerung vollständig geimpft sind und Impfstoffe Krankenhausaufenthalte um 90 % reduzieren, würden wir erwarten, dass 2/3 der Krankenhausaufenthalte auf geimpfte Personen entfallen: https://t.co/rMln0oncu2
#LongCovid wird immer noch unterschätzt. Eine andauernde Fehlfunktion der kleinen Gefässe und eine Unterdurchblutung bestimmter Gehirnbereiche sind nur ein Beispiel. Auch 20% der Geimpften, die Durchbruchinfektion haben, könnten betroffen sein. https://t.co/WkhOROqs34
Wir sollten die Inzidenz niedrig halten. Weil im Herbst der Schutz vor Ansteckung und vor LongCovid verloren geht. Wahrscheinlich schützt Impfung nur noch 50% vor Ansteckung. Und 20% der Angesteckten nach Impfung bekommen LongCovid. https://t.co/CXw497lzTG
Oh Mann. Wenn man schon @chrischirp zitiert, die mit der ganzen #indieSAGE-Mannschaft sehr auf der #NoCovid Welle reitet und dann noch Fantasieraten von #Geimpften mit #LongCovid hinzuphantasiert… Und das lesen jetzt 570.000 Leute und glauben womöglich das seien Fakten.
Die Studie ist relevant. Sie zeigt, dass Kinder unter 3 J bei Delta Variante besonders ansteckend sind, mehr als ältere Kinder. Daher sollten sich Eltern auf jeden Fall impfen lassen. https://t.co/VldoT42Zht
Führen zu mehr Haushaltsinfektionen als ältere Kinder. Vermutlich weil sie mehr Kontakt haben (logisch, sind ja kranke Kleinkinder). Kinder immer noch weniger ansteckend als Erwachsene (siehe conclusion). Und: es geht nicht um Delta, sondern um die zweite Welle Ende 2020.
(1) Diese interessante Studie aus Irland zu #LongCovid weist erneut darauf hin, dass die Blutgerinnung und die kleinsten Gefässe dabei beschädigt werden. Das würde die Denkstörungen und die Minderdurchblutung des Gehirns bei vielen LC Patienten erklären https://t.co/kKa8tOMm27
Es zeichnet sich ab, dass BionTech Impfung Wirkung gegen Ansteckung relativ schnell verliert, schneller wohl als AstraZeneca. Der Schutz gegen schwere Krankheit hält länger an, aber nicht für Risikopatienten. Daher müssen wir sie vor dem Winter impfen. https://t.co/SuRzSQjCrJ
Diese Studie an Makaken Affen zeigt, dass die Dosis SarsCoV2 beim einatmen 5x höher ist für eine Infektion mit Fieber als für fieberfreie Infektion. Ein sehr starkes Argument für das Tragen von Masken. https://t.co/8t30IQjYoX
Es ist erstaunlich. In den US appellieren öffentlich führende Kinderärzte jetzt an Regierung für besseren Covid Schutz der Kinder. Sie haben Angst vor Langzeitschäden. Bei uns beginnt eine stille Durchseuchung. In Hospitalisierungsquote spielt #LongCovid für Kinder keine Rolle https://t.co/L2fJFi9gNj
Diese wichtige UK Studie zeigt, dass das Risiko für #LongCovid bei Infizierten nach Impfung deutlich geringer ist. Impfung schützt daher vor langfristigen Schäden und Infektion. Ein wichtiger neuer Grund für eine Impfung. https://t.co/JyChlbsJRv
In Japan festgestellte Mu Variante ist gegen BionTech oder Antikörper Genesener resistenter als ALLE anderen Varianten. Wenn eine Variante wie Mu auch zu hoher Ansteckung mutiert wäre das äusserst gefährlich. Bei Milliarden Ungeimpfter jederzeit möglich. https://t.co/1GO5utajhR
Wichtige Studie. Sie zeigt, dass in China Frauen nach Covid, auch bei leichterem Verlauf, später hormonell weniger fruchtbar waren. Ob sich Fruchtbarkeit erholt ist unklar. Somit gilt: nicht die Impfung bedroht die Fruchtbarkeit sondern die Infektion https://t.co/hvs9huy1XJ
Studie zeigt erneut, dass Covid im Gehirn Spuren hinterlässt, auch bei relativ leichten Fällen. Da Veränderungen Gehirnbereiche beschädigen, die bei Alzheimer beschädigt sind, können wir alle nur hoffen, dass die Veränderungen weggehen. Niemand weiss das https://t.co/h6VaEglyUx
1/ Ich versuch mal mein Glück: Es handelt sich um eine Studie, die automatisiert MRT-Aufnahmen von Menschen vor und nach #COVID-Infektion verglichen hat, gepaart mit einer Kontrollgruppe ohne C19-Infektion. Die Probanden wurden zudem neuropsychologisch getestet. https://t.co/8vIxiXqjdi
Moderna hat leicht höheres Risiko von Herzmuskelentzündungen als BionTech, besonders bei jungen Männern. Moderna ist höher dosiert als BionTech, hat daher auch wahrscheinlich einen etwas längeren Schutz. Die Entscheidung in Schweden/Dänemark ist fragwürdig https://t.co/2cFsWJR7B6
Oder es ist vorsichtiger. Moderna hat einen wahrscheinlich längeren Schutz vor „any infection", vor schweren Verläufen, Hospitalisation und Tod helfen beide Impfstoffe und auch beide länger als 6 Monate lieber @Karl_Lauterbach. https://t.co/njwQKQfMRW
Die Daten stammen aus den zusammengelegten Gesundheitsdatenbanken von 🇩🇰 , 🇫🇮, 🇳🇴& 🇸🇪 . So etwas wirkt aus deutscher Sicht möglicherweise unwirklich futuristisch, ist allerdings alles andere als „fragwürdig“.
Hier sieht man die neuesten Daten aus UK zu Covid. Die Aufarbeitung ist erneut sehr gut. Die Hospitalisierungen der Kinder sind problematisch trotz der Tatsache, dass kaum Kinder sterben. Man muss von bedeutsamer Quote #LongCovid bei Kindern ausgehen. https://t.co/U601mIozUSpic.twitter.com/jBwhe5h6FP
Es sind Tweets wie dieser, der heutige mit den ONS-Daten, die ganz sicher keine hohe Quote von #LongCovidKids zeigen, der mit der nebenwirkungsfreien Impfung vor einiger Zeit oder der mit der noblen Idee #NoCovid warum man Ihnen Populismus vorwerfen muss. https://t.co/i1YJ9CMw2N
Methodisch sehr hochwertige Studie Oxford Universität zur Wirksamkeit Lockdown Massnahmen in 2. und 3. Welle. Die Massnahmen verloren leicht an Wirkung im Vgl. zur 1. Welle, blieben aber sehr wirksam. So erklärt sich der relativ grosse Erfolg Deutschlands https://t.co/PaOBehyE33
(1) Immer wieder wird Kindern in Deutschland abgesprochen, dass auch sie stark unter #LongCovid leiden. Das ist im Ausland anders. In Israel wurden Eltern von Covid Kindern befragt. 6 Mon nach Infekt hatten je nach Alter noch 11% Symptome. https://t.co/mUK4N1vJtm
Für die vielen, die Covid überstanden haben, muss noch viel getan werden. Studien zeigen klar, dass die Krankheit den Prozess der Alterung deutlich beschleunigt. Man altert im Zeitraffer und wird gegen chronische Krankheiten anfälliger sein. https://t.co/x4ZlgBK6Cp
Same procedure at nearly every day: Nein, man altert nicht im Zeitraffer und das steht in der Arbeit auch so nicht drin. Auch nicht, wenn man die Ergebnisse stark verkürzt, @Karl_Lauterbach, sondern ⬇️ https://t.co/CjfD9c33Gl
In den Niederlanden steigt nach Lockerungen die Zahl der Corona-Infektionen stark an, bei einer mit unserer vergleichbaren Impfquote. Auch die Zahl der Todesfälle ist relativ hoch. Ob wir den FreedomDay von Gassen und Spahn bekommen steht noch offen. https://t.co/PsifNXnJIQ
Die Fallzahlen steigen wieder, dass war zu erwarten. Die Impfquote ist noch zu niedrig. Die FreedomDay Diskussion hilft auch nicht. Mehr 2G und eine neue, innovativere Impfquote würden helfen. https://t.co/rG5gJFRkVn
Wir führen jetzt eine innovative Impfquote nach Schroedinger ein. Sie ist gleichzeitig hoch genug und zu niedrig. Im Winter öffnen wir den Kasten und schauen nach. https://t.co/kAwAWA4D1z
Diese wichtige Arbeit zu Covid von der Uni Lübeck bestätigt die Hypothese, dass SARSCoV2 im Gehirn die kleinen Blutgefässe zerstört. Das kann viele neurologische Covid Befunde erklären. Obwohl die Studie dies nicht zeigt legt das Ergebnis ein erhöhtes späteres Demenzrisiko nahe https://t.co/ncVV9wTWW2
Die Studie zeigt keine Hinweise auf ein späteres erhöhtes Demenzrisikio, untersucht es auch nicht und die Autoren spekulieren auch nicht darüber. Es ist eine extrem komplexe Grundlagenforschungsstudie, bei der es im wesentlichen um die Frage geht, ist #COVID19 eine Gefäßkrankheit https://t.co/RvSBBIifIU
Diese grosse Studie der Uni Dresden in Kooperation mit RKI und Krankenkassen zeigt, dass Kinder so viel #LongCovid in Deutschland erlitten wie Erwachsene. Der Satz: Kinder werden bei Covid nicht ernsthaft krank, entspricht nicht den Studien preprint_post_covid_2021-10-20.pdf  pic.twitter.com/KUoz8H3t09
… und die wichtigste Erwiderung darauf: https://t.co/JqodUte4iY Und jetzt würden wir uns auch noch auf den korrekten Link zur Studie freuen, @Karl_Lauterbach, damit man sie auch lesen kann …
Herr @Karl_Lauterbach so langsam wird das was sie betreiben Desinformation! Selbst in der Zusammenfassung steht schon dass die Rate bei Kindern niedriger ist! Long Covid ist ein wichtiges Problem, aber das was sie hier betreiben ist schandhaft! https://t.co/ePDlpCOL9w
Cambridge Uni Studie zeigt, wie andere Studien zuvor, dass #LongCovid mit Verlusten der Denkfähigkeit einhergehen kann. Andere Studien haben Verlust an grauer Substanz gezeigt. Das sind bei und immer noch Tabuthemen https://t.co/K4VthFBDmG
Der Freedom Day in Dänemark ist komplett gescheitert. Ich erinnere mich noch als Herr Gassen, KBV, das gleiche bei uns forderte und empört war, als ich das im Streitgespräch mit ihm ablehnte. https://t.co/ZUTwCGVkXl
Wenn man komplexe Themen auf simple Phrasen runterbricht & behauptet, der Freedom Day in Dänemark wäre gescheitert, dann könnte man genauso behaupten, dass der Nicht-Freedom Day in Deutschland noch deutlicher gescheitert ist.
Das Paper hatten wir ähnlich unglücklich kommuniziert weiter oben schon mal:
1) Immer wieder wird bestritten, dass es #LongCovid überhaupt gibt. Daher hier noch einmal eine besonders wichtige Studie der Oxford Uni zur Klarstellung. Die Studie konnte besonders genau die Gehirne von Covid Patienten mit Gesunden vergleichen. Ergebnis: https://t.co/nLI1IOdA0m
Dass Covid den Gehirnstoffwechsel beschädigen kann, auch bei leichteren Verläufen, ist lange bekannt. Das will in Deutschland kaum jemand hören. Konzept ist „tot oder genesen“. Aber bei Kindern sollten wir so fair sein, jede unnötige Infektion zu vermeiden https://t.co/E0LhXlqxSg
Im nächsten Jahr werden hunderttausende in Deutschland mit den Folgen von #LongCovid kämpfen. Sie werden nicht arbeiten. Auch dieser wirtschaftliche Schaden zählt. Er ist grösser, als 2 Wochen die Geschäfte zu schliessen. https://t.co/AqSrCOlUKz
Hier noch kleine Zusammenfassung, worum es in dem Paper wirklich geht:
Es ist eine australische prospektive Studie, bei der 253 Patienten, die an #EBV, einer bakteriellen Infektion mit Coxiella burnetii oder einer Virusinfektion mit Ross River-Virus (das kannte ich gar nicht) https://t.co/RxHAUWZm1B
Das ist der bisher beste Thread zu Omicron. In der Quintessenz wissen wir derzeit 1) dass Omicron sehr gefährlich ist und 2) dass es noch immer unklar ist ob die Gefahr auch für Geimpfte wirklich grösser ist. https://t.co/FmpOOjQwJb
Lieber @Karl_Lauterbach. Dies ist ein wunderbarer wissenschaftlicher Thread, nicht die übliche banale Panikmache eines @DrEricDing. Also geben Sie sich bitte etwas mehr Mühe beim Zusammenfassen.
Ja, ich hatte erst gerade geschrieben, dass so langsam Schluss mit den Corona-Beiträgen sein soll (Link) und jetzt kommt doch noch einer. Karl Lauterbach hat mal wieder getwittert und zwar das hier:
Ein typischer Lauterbach, denn „Obwohl die Studie dies nicht zeigt legt das Ergebnis ein erhöhtes späteres Demenzrisiko nahe“ steht da geschrieben. Doch darum geht es in der Studie gar nicht. Und das triggert mich so an, dass ich jetzt doch noch mal was dazu schreiben muss.
Das Paper
Es geht um dieses Paper:
Wenzel, J., Lampe, J., Müller-Fielitz, H., Schuster, R., Zille, M., Müller, K., Krohn, M., Körbelin, J., Zhang, L., Özorhan, Ü., Neve, V., Wagner, J. U. G., Bojkova, D., Shumliakivska, M., Jiang, Y., Fähnrich, A., Ott, F., Sencio, V., Robil, C., … Schwaninger, M. (2021). The SARS-CoV-2 main protease Mpro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nature Neuroscience, 394. https://doi.org/10.1038/s41593-021-00926-1
Die Autoren haben selber einen kleinen Twitter-Thread hierzu erstellt:
Our finding on #SARSCoV2 and the brain´s microvasculature was published in @NatureNeuro today. We used cells, animal models, and #COVID19 patients samples to show that the virus kills brain endothelial cells, possibly explaining neurological symptoms: https://t.co/ZV9SHqef8V.
Ich habe hier ein paar mehr Zeichen zur Verfügung, daher versuche ich auch noch mal mein Glück beim Erklären des Papers, denn es berührt eine ganz spannende Frage, nämlich die, ob die Behauptung, die man oft so lapidar hört „COVID-19 ist eine Erkrankung der kleinen Gefäße“ stimmt. Hierzu gibt es nämlich bislang unterschiedliche Auffassungen und auch hochrangig publizierte Zweifel (z.B. McCracken et al.). Und die Arbeit schneidet am Rand die ebenso wichtige Frage an, sind die Erkenntnisse der Autoren COVID-exklusiv oder werden hier allgemeingültige Mechanismen einer Virusinfektion aufgedeckt.
Das Paper ist lang (in der pdf-Version mit Methodik-Anhang 29 Seiten) und komplex, da es eine Arbeit aus der Grundlagenforschung ist, bei der es um molekularbiologische Mechanismen bei einer COVID-19-Infektion geht. Also keine leichte Kost und damit eigentlich auch nur bedingt Twitter-kompatibel.
Die Fragestellung
Grundlage der Studie sind Erkenntnisse, dass bei schwer betroffenen COVID-19-Patienten vermehrt ischämische Schlaganfälle, sowie epileptische Anfälle und Enzephalopathien (oft in der Form eines Delirs) – in der akuten und post-akuten Phase – auftreten. Zudem nehmen die Autoren MRT-Aufnahmen von COVID-19-Erkrankten, die Mikroangiopathie-, bzw. Vaskulitis-ähnliche Läsionen zeigten als Grundlage ihrer Überlegungen.
Methodik der Studie
Ist COVID eine Kapillarerkrankung?
Die Autoren haben sich ganz verschiedener Indizien bedient, sie haben Gehirn-Proben aus Autopsiestudien von 17 an COVID-19-Verstorbenen untersucht und dies mit einer Kontrollgruppe von 23 aus anderen Gründen verstorbener Probanden und zwei Tiermodellen (Maus und Hamster) verglichen. In den Proben Verstorbener suchten sie nach Resten von kleinen Gefäßen, vornehmlich Kapillaren und fanden dünne röhrenartige Strukturen, sogenannte „string vessels“, bei denen offenbar nur noch die Basalmembran (Link Wikipedia) übrig geblieben, das Endothel (Link Wikipedia) hingegen untergegangen war.
A thread: Having clinical data in mind that hints at microvascular changes in COVID-19 patients, we started to look into the microvasculature of brain samples of deceased COVID-19 patients. We found an increased amount of dead capillaries, so-called string vessels. pic.twitter.com/kD22IRg83t
Bei den COVID-Toten fanden sich deutlich mehr string vessels als in der Kontrollgruppe, auch im Vergleich zu beatmeten, nicht-COVID-Patienten, so dass die Autoren eine systemische Hypoxie als Grund des Kapillarverlustes unwahrscheinlich ansehen. Zur Bestätigung ihrer Hypothese untersuchten sie das Auftreten eines Proteins, welches bei einem Zelltod (Apoptose) vermehrt auftritt, der Caspase-3. Dieses fand sich ingesamt selten, aber öfter bei an COVID-Verstorbenen als in der Kontrollgruppe. Auch bei mit COVID-infizierten Hamstern konnten vermehrte string vessels ab dem vierten Tag nach Infektion nachgewiesen werden, ebenso bei transgenen Mäusen.
In einem nächsten Schritt haben die Autoren untersucht, ob SARS-CoV-2 überhaupt in der Lage ist, Endothelzellen im Gehirn zu infizieren. Dafür färbten sie verschiedene – in der Vergangenheit mit einer SARS-CoV-2-Infektion assoziierte – Oberflächeneiweiße im Mausmodell ein, nämlich ACE2 (siehe auch Blogbeitrag zum Thema COVID und Neurodegeneration), Neuropilin-1 (was als Nrp1 abgekürzt wird) und Basigin (BSG). ACE-2 wurde hauptsächlich vom die Kapillaren umgebenden Bindegewebe exprimiert und eher wenig von den Kapillaren, anders Nrp1 und BSG, die sich vor allem in den Endothelzellen fanden. Dies war auch im Hirngewebe Verstorbener so. Allerdings hat dieses Vorgehen einen Schönheitsfehler, da ACE-2 mit versiegendem Blutstrom (und das ist sowohl bei toten Mäusen, als auch toten Menschen so) rasch weniger exprimiert wird. Glich man das mit einem Labortrick aus, konnte man aber ACE-2 auf Endothelzellen und sogar Spike-Protein in den Zellen nachweisen.
Was macht SARS-CoV-2 mit den infizierten Zellen?
Um zu Überleben und vervielfältigt zu werden, kann SARS-CoV-2 die infizierten Zellen verändern („manipulieren“). Dafür spaltet es ein Protein mit dem schönen Namen NEMO (was für nuclear factor (NF)-κB essential modulator steht). NEMO aktiviert dem Namen nach NF-κB (Link Wikipedia), was wiederum eines der zentralen Proteine der zellulären Immunantwort, von und für Entstehungen von Entzündungen und der Einleitung von Apoptose-Vorgängen. SARS-CoV-2 bildet zwei Schneideproteine, sogenannte Proteasen, u.a. Mpro. Mpro kann NEMO zerschneiden, so dass es nicht mehr funktioniert. In der Folge fehlt eine IL-1-vermittelte Entzündungsreaktion, so dass das Virus nicht bekämpft wird. Dummerweise ist NEMO auch für die Funktionsfähigkeit von Endothelzellen verantwortlich. Zellen, in den mit MproNEMO zerschnitten wurde, gingen öfter in den Zelltod, als normal. Kapillaren scheinen dies wiederum öfters als andere Zellen zu machen. Durch den Tod vieler Kapillaren wurde im Tiermodell dann die Blut-Hirn-Schranke durchlässiger. Die Dichte der Perizyten veränderte sich nur leicht, Mikroglia hingegen wurde aktiviert. In ihren Untersuchungen konnten die Autoren auch erhöhte Konzentrationen von saurem Gliafaserprotein (GFAP) nachweisen, was kongruent zu anderen Arbeiten ist, wie hier im Long Covid-Beitrag erwähnt.
Ein therapeutischer Ansatz
In einem letzten Schritt untersuchten die Autoren Wege, diesen Prozess aufzuhalten und stießen auf ein weiteres Eiweiß mit dem Namen RIPK3. RIPK3 spielt ebenfalls im Zusammenspiel mit NF-κB eine Rolle in der Induktion von Apoptose-Vorgängen. In einem Hamstermodell führte ein RIPK-3-Mangel zu einer Rückbildung der oben beschriebenen Auffälligkeiten und verbesserte auch das Überleben der Hamster. Allerdings können RIPK3-Inhibitoren selber Zelltode auslösen, so dass man sich zur Untersuchung eines vorgelagerten Signalweges mit einem Protein namens RIPK1 zuwandte, was unkomplizierter beeinflussbar ist.
In a final experiment, we treated mice that express the SARS-CoV-2 main protease in the brain endothelium with a necroptosis inhibitor and found, indeed, the microvascular pathology to be improved. pic.twitter.com/MnlyVG3keg
Die Autoren sehen in ihrer Studie einen Ansatz zur Erklärung vaskulärer Komplikationen bei COVID-Erkrankungen, aber auch von Long Covid-Beschwerden. Das Außer-Kraft-Setzen von NEMO ist für verschiedene andere Coronaviren beschrieben, aber auch für Influenza-Viren (siehe Häfner und Wei et al.) und für onkologische Erkrankungen.
Nach meinem Dafürhalten spricht das dafür, dass der beschriebene Pathomechanismus zumindest in dem Teil der ohne ACE-2-Rezeptor auskommt eher ein genereller Mechanismus sein dürfte, als ein COVID-spezifischer.
Dennoch können die Autoren den Pathomechanismus kleinteilig beschreiben und belegen und zeigen zudem mit RIPK1-Antagonisten einen Ansatz einer spezifischen Anti-COVID-Behandlung.
Wo man weiterlesen kann
McCracken, I. R., Saginc, G., He, L., Huseynov, A., Daniels, A., Fletcher, S., Peghaire, C., Kalna, V., Andaloussi-Mäe, M., Muhl, L., Craig, N. M., Griffiths, S. J., Haas, J. G., Tait-Burkard, C., Lendahl, U., Birdsey, G. M., Betsholtz, C., Noseda, M., Baker, A. H., & Randi, A. M. (2021). Lack of Evidence of Angiotensin-Converting Enzyme 2 Expression and Replicative Infection by SARS-CoV-2 in Human Endothelial Cells. Circulation, 143(8), 865–868. https://doi.org/10.1161/CIRCULATIONAHA.120.052824
Wei, F., Jiang, Z., Sun, H., Pu, J., Sun, Y., Wang, M., Tong, Q., Bi, Y., Ma, X., Gao, G. F., & Liu, J. (2019). Induction of PGRN by influenza virus inhibits the antiviral immune responses through downregulation of type I interferons signaling. PLOS Pathogens, 15(10), e1008062. https://doi.org/10.1371/journal.ppat.1008062
Wenzel, J., Lampe, J., Müller-Fielitz, H., Schuster, R., Zille, M., Müller, K., Krohn, M., Körbelin, J., Zhang, L., Özorhan, Ü., Neve, V., Wagner, J. U. G., Bojkova, D., Shumliakivska, M., Jiang, Y., Fähnrich, A., Ott, F., Sencio, V., Robil, C., … Schwaninger, M. (2021). The SARS-CoV-2 main protease Mpro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nature Neuroscience, 394. https://doi.org/10.1038/s41593-021-00926-1
In Teil 1 der Reihe ging es um die Grundlagen zum Thema Neurotropie von SARS-CoV-2 und um die Frage, wie das Virus überhaupt ins ZNS kommen und wie man es dort nachweisen kann. In Teil 2 bin ich dem Thema nachgegangen, ob und wie SARS-CoV-2 vielleicht Trigger von Neurodegeneration sein kann. Ich hatte mich in den ersten beiden Teilen der Reihe mit Wertungen und Einsortierungen von Sachverhalten sehr zurückgehalten. Das will ich jetzt hier tun und wenn Ihr das anders seht, dürft Ihr gern Teil 1 und 2 lesen und Euch Eure eigene Meinung bilden und diese vertreten.
Ich möchte im wesentlichen drei Fragen für mich beantworten, legen wir also los:
Was sind Besonderheiten von SARS-CoV-2 und wo decken wir gerade allgemeingültige Mechanismen auf?
Das ist für mich eine sehr spannende Frage, die ich ja schon mehrfach, u.a. kurz auf der Startseite, skizziert habe. Der Trend in den populärwissenschaftlichen Medien geht sehr zur Attribuierung von vermeintlichen exklusiven Mechanismen von SARS-CoV-2. Drama und Alarm verkaufen sich halt besser. Der Trend in den wissenschaftlichen Papern aber auch, aber das muss man etwas relativieren. Jeder, der regelmäßig Paper liest kennt das Phänomen, dass die Ergebnisse im Abstract und in der Diskussion am Ende des Beitrags in der Regel prägnant bis dramatisierend dargestellt werden. Das ist ja auch kein Wunder, weil wenn man schreiben würde: Ja, unsere Forschungsergebnisse sind nicht so berauschend, die Datenlage dünn und so richtig beweisen können wir unsere Arbeitshypothese auch nicht, würde es schwer mit der Veröffentlichung. Deshalb sind ja oft die Absätze über die Limitationen einer Studie viel interessanter als die (vermeintlich) eindeutigen und überzeugend dargestellten Studienergebnisse. Ein Beispiel hierfür kann die im zweiten Teil erwähnte FDG-PET-Studie bei sieben Kindern sein (Morand et al.), bei der nur bei dreien überhaupt ein positiver COVID-Nachweis vorlag, so dass die durchaus berechtigte Frage gestellt werden kann, was die Arbeit überhaupt aussagen kann.
Wenn ich das richtig sehe, dann gibt es einen Mechanismus, der sehr exklusiv für SARS-CoV-2 (und für SARS-1, nicht aber für andere Corona-Viren und auch nicht für andere Atemwegserkrankungs-Viren) ist und das ist die Bindung an ACE-2 in menschlichen Zellen. Über diesen Mechanismus lassen sich nahezu alle Krankheitsmanifestationen erklären, die wir bei COVID-19 sehen.
Und wenn ich es auch richtig sehe, dann gibt es ganz viele Dinge, die überhaupt nicht exklusiv für SARS-CoV-2 sind, weil es andere Viren und v.a. andere Atemwegserkrankungs-Viren auch genau so machen: Die Infektion des ZNS über die Riechzellen, die Aktivierung von Mikroglia und kognitive Defizite nach der Infektion. Viele Viren stehen seit Jahren, und Influenza seit eigentlich einem Jahrhundert in Verdacht (vgl. hier) neurodegenerative Erkrankungen auslösen oder triggern zu können.
Aktuell werden mit riesigem personellen und finanziellen Aufwand Dinge beleuchtet, die vermutlich eher allgemeingültige Mechanismen darstellen dürften. Und es werden Dinge untersucht, die bislang so noch niemand untersucht hat. Studien wie die aus dem UKE zu kognitiven Defiziten auch nach leichten COVID-Verläufen (Woo et al.) gibt es meines Wissens nicht (bzw. in dieser Qualität nicht) zu anderen häufigen Infektionserkrankungen. Und Studien wie die UK Biobank-Studie (Douaud et al.) erst Recht nicht.
Sollten uns die Erkenntnisse besorgen?
Ich denke nicht. Und zwar aus Gründen. Es werden ja im Endeffekt zwei Hauptsorgen immer wieder thematisiert: Die Triggerung von neurodegenerativen Prozessen und eine Neuroinvasion mit Spätschäden ähnlich Polio oder der SSPE bei Masern.
Bei dem Thema neurodegenerative Erkrankungen nach Infektion muss man den Satz von gerade noch mal betonen: Bei der Influenza vermuten wir seit der spanischen Grippe, dass Influenza Parkinson-Erkrankungen auslösen kann. Gestört hat es uns nur nie. Und ganz viele, die das Thema Neurodegeneration durch SARS-CoV-2 jetzt immer und immer wieder laut wiederholen haben sich bis vor zwei Jahren nie gegen Grippe impfen lassen, „weil das ja nur der Pharma-Industrie nützt“. Dazu kommt, dass sich hier seit Jahren bis Jahrzehnten wissenschaftlich nichts tut. Gerade bei Parkinson mit dem Beginn der Neurodegeneration im Riechhirn und im Darm-Nerven-System ist ein auslösender Umweltfaktor, den wir einatmen oder runterschlucken extrem wahrscheinlich, viel wahrscheinlicher als bei der Ausbreitungsmorphologie von Tau und ß-Amyloid bei der Alzheimer-Demenz. Nur, wir finden diesen Umweltfaktor bislang nicht (vermutlich sind es aber auch mehrere). Auch die Viren der Herpes-Gruppe stehen seit Jahrzehnten in Verdacht an Neurodegeneration beteiligt zu sein. Und die sind wirklich neurotrop, neuroinvasiv und neuropathogen und dazu noch DNA-Viren, die sogar unser Erbgut verändern. Aber bei EBV haben wir eine Durchsuchung (in diesem Kontext wäre der Begriff richtig benutzt) von 80-95% bei jungen Erwachsenen, bei VZV von 95%, bei HSV von 90%. Das macht die kausale Zuordnung zu den ebenfalls häufigen, aber nicht derart häufigen neurodegenerativen Erkrankungen schwer. Wenn uns das alles gar nicht stört bislang – und ich kann den Gedanken nachvollziehen, da wir uns eh alle mit diesen Viren infizieren – warum sollten wir dann bei COVID-19 anders reagieren?
Interessant ist auch die Sorge vor COVID-Spätschäden analog zu Polio oder der SSPE nach Maserninfektion. Im Gegensatz zu diesen Erkrankungen gelingt bei COVID-19 der PCR-Nachweis im Liquor nur in ca. 6% und ein spezifischer intrathekaler Antikörper-Nachweis bei 23% der Infizierten. Und in erster Linie kann dann Antikörper oder Virus-RNA nachgewiesen werden, wenn die Patienten einen schweren Krankheitsverlauf mit ernsthaften neurologischen Komplikationen erleiden. Bei einer mild verlaufenden COVID-19-Infektion ohne neurologische Komplikationen gibt es so gut wie keinen reproduzierbaren COVID-Nachweis im Liquor. Dass ein nicht mehr nachweisbares Virus dann spezifische Spätschäden machen soll, auch nach leichten Krankheitsverläufen, ist nicht plausibel und es ist bei den zitierten anderen Krankheiten eben auch nicht so.
Was erhoffe ich mir von der COVID-Forschung?
Ich persönlich sehe die Chance durch den unglaublichen Fokus auf COVID-19 und seine Folgen bislang un- oder nur bruchstückhaft verstandene allgemeingültige Krankheitsmechanismen von Infektionskrankheiten entschlüsseln zu können, beim Thema Neurodegeneration ihren Einfluss auf die Kognition und vielleicht ja wirklich auch mal auf das Thema Triggerung neurodegenerativer Prozesse. Und beim Long Covid-Syndrom Licht in das Thema chronisches Erschöpfungssyndrom zu bekommen, denn da sind der aktuelle Wissenstand und die verhärteten Fronten zwischen CSF-Befürwortern und -Skeptikern eine Katastrophe. Gelänge das, wären das extrem große Errungenschaften.
Literaturangaben
Morand, A., Campion, J. Y., Lepine, A., Bosdure, E., Luciani, L., Cammilleri, S., Chabrol, B., & Guedj, E. (2021). Similar patterns of 18F-FDG brain PET hypometabolism in paediatric and adult patients with long COVID: a paediatric case series. European Journal of Nuclear Medicine and Molecular Imaging, 0123456789. https://doi.org/10.1007/s00259-021-05528-4
Woo, M. S., Malsy, J., Pöttgen, J., Seddiq Zai, S., Ufer, F., Hadjilaou, A., Schmiedel, S., Addo, M. M., Gerloff, C., Heesen, C., Schulze Zur Wiesch, J., & Friese, M. A. (2020). Frequent neurocognitive deficits after recovery from mild COVID-19. Brain Communications, 2(2), 1–9. https://doi.org/10.1093/braincomms/fcaa205
Douaud, G., Lee, S., Alfaro-Almagro, F., Arthofer, C., Wang, C., Lange, F., Andersson, J. L. R., Griffanti, L., Duff, E., Jbabdi, S., Taschler, B., Winkler, A., Nichols, T. E., Collins, R., Matthews, P. M., Allen, N., Miller, K. L., & Smith, S. M. (2021). Brain imaging before and after COVID-19 in UK Biobank. MedRxiv : The Preprint Server for Health Sciences. https://doi.org/10.1101/2021.06.11.21258690
In der Vergangenheit hatte ich Diskussionen mitbekommen (und mich auch in welche verstrickt), on SARS-CoV-2 nun total, nicht sonderlich oder nur ein bisschen neurotrop sei. Aber eigentlich ist das eine total behämmerte Frage. Wir kennen Viren, die hauptsächlich das Nervensystem infizieren und dort persistieren, wie die Herpes-Viren, wir kennen Viren, die ausschließlich das Nervensystem befallen, wie das JC-Virus und wir kennen die große Gruppe der Viren, die relativ wahllos Atemwegs-Schleimhaut, Lungen, Magen-Darm-Schleimhaut und auch das ZNS infizieren können, wie die meisten Atemwegserkrankungs-Viren und Gastroenteritis-Viren. Und in diese Gruppe gehören auch die Coronaviren und damit auch SARS-CoV-2.
Alle in der Neurologie Tätigen kennen die typischen Fälle (vermeintlich) viraler Meningitiden und Enzephalitiden, bei denen nie ein Erregernachweis gelingt und man immer heilfroh ist, wenn die Patienten dann Kinder im Kita- oder Grundschulalter zu Hause haben, die „auch gerade einen Infekt“ hatten. Und ja, manchmal gelingt uns der Nachweis von Entero- oder Adenoviren (und ganz selten von Coxsackie-Viren) und häufig nicht.
Wie SARS-CoV-2 in das ZNS gelangen kann
Zwei gut lesbare und interessante Arbeiten sind die Revier-Paper von Fotuhi et al. und von Yachou et al., welche aber aus 2020 stammen, was man an manchen Stellen merkt. Die Autoren arbeiten die Wege auf, wie SARS-CoV-2 überhaupt ins ZNS gelangen kann. Eine direkte Infektion des ZNS auf dem Blutweg ist nämlich für die meisten Viren gar nicht so banal möglich, da die Blut-Hirn-Schranke nicht ohne weiteres überwunden werden kann. Ein häufig benutzter Weg ist daher die Infektion der Riechschleimhaut, der Nasen-Rachen-, Darmschleimhaut oder des Plexus choroideus, um in das ZNS zu gelangen. Diese Mechanismen sind für viele Viren beschrieben, auch und insbesondere für andere Atemwegserkrankungserreger wie RSV und Influenza, aber auch für verschiedene Corona-Viren (sowohl tierische, als auch humanpathogene Stämme wie SARS-1, MERS und SARS-CoV-2). Von dort bedienen sich viele Viren des retrograden axonalen Transports, mit dem sie vom Infektionsort Richtung Nervenzelle transportiert werden. Das kennen alle Mediziner von der Tollwut und den Herpesviren, es ist aber ein relativ allgemeiner und häufig benutzter Infektionsmechanismus. Neben dem Riechnervenbündel betrifft das häufig den N. trigeminus und die sensiblen Anteile des N. vagus. Im Nervensystem angekommen (dem Hirnstamm, den hier befinden sich die Hirnnervenkerne), treffen sie auf die verschiedenen Abwehrmechanismen des Körpers. Eine der ersten Strukturen, auf die Viren dann treffen und die aktiviert wird, ist die Mikroglia. Das sind mononukleäre Fresszellen, die Aktivierung von Mikroglia spricht immer für eine Verletzung des ZNS oder einen informatorischen Prozess. Durch die Mikrogliaaktivierung kommt es zur Ausschüttung von Zytokinen und dem Anlocken von weiteren Immunzellen. Ausgeprägte Entzündungsprozesse des ZNS gelten jedoch als Aktivator von neurodegenerativen Prozessen, vermutlich spielt auch hier die Mikroglia eine entscheidende Rolle .
Wo man weiterlesen kann
Fotuhi, M., Mian, A., Meysami, S., & Raji, C. A. (2020). Neurobiology of COVID-19. Journal of Alzheimer’s Disease, 76(1), 3–19. https://doi.org/10.3233/JAD-200581
Yachou, Y., El Idrissi, A., Belapasov, V., & Ait Benali, S. (2020). Neuroinvasion, neurotropic, and neuroinflammatory events of SARS-CoV-2: understanding the neurological manifestations in COVID-19 patients. Neurological Sciences, 41(10), 2657–2669. https://doi.org/10.1007/s10072-020-04575-3
Wie kann man Neurotropie und Neuroinvasion messen?
Kurzer Ausflug in die Grundlagen der Liquordiagnostik
In der Neurologie sind heutzutage zwei Nachweis-Wege einer Infektion des ZNS üblich, wobei der Nachweis in der Regel aus dem Liquor per Liquorpunktion geführt wird: Der direkte Erregernachweis per PCR und der Nachweis spezifischer Antikörper gegen den Erreger, welche im ZNS in höherer Konzentration vorliegen als im Blut. Finden sich Antikörper gegen einen Erreger im Liquor stellt sich die Frage, ob diese wirklich im ZNS produziert wurden, oder aus dem Blut in den Liquor hinübergeschwappt sind. Ob und in welchem Maß das passiert, hängt mit der Dichtigkeit der Blut-Hirn-Schranke zusammen. Diese ist bei jungen und gesunden Menschen sehr dicht, im Alter zunehmend undichter und bei entzündlichen Prozessen oft sehr durchlässig. Je durchlässiger die Blut-Hirn-Schranke aber ist, desto mehr Antikörper aus dem Blut werden sich im Liquor finden. Mit dem Reiber-Schema wird versucht, diesen Sachverhalt zu berücksichtigen. Die Idee ist, ein nur in der Leber synthetisiertes möglichst kleines Protein, nämlich Albumin zu nehmen. Mit dem kann dann das Maß der Dichtigkeit der Blut-Hirn-Schranke bestimmt werden, da alles Albumin, was sich im Liquor findet unweigerlich aus dem Blut, bzw. aus der Leber stammen muss. Viel Albumin im Liquor bedeutet dann eine undichte Blut-Hirn-Schranke, wenig eine sehr dichte. Setzt man nun die verschiedenen Antikörper in das Verhältnis des Albumins im Serum zum Liquor, so kann man ableiten, wie viel Antikörper intrathekal, also im Liquor produziert wurden und wie viel rübergeschwappt sind. Dieses Verhältnis der Antikörper von Liquor zu Serum nennt man Antikörper-Spezifitäts-Index (ASI). Für die meisten Erkrankungen wird ein ASI von > 1,5 (also 1,5 mal so viele Antikörper im Liquor wie im Serum) gefordert, um eine sichere intrathekale Antikörperproduktion anzunehmen zu können.
Ich werde das Thema Reiber-Schema auch noch mal im Blog aufarbeiten, fürs Erste verweise ich aber mal auf die Webseite von Herrn Reiber himself: Link.
Erreger, die man nicht im Liquor per PCR und/oder Antikörper-Wert messen kann, obwohl sie da und auch weiterhin infektiös sind, kommen wirklich selten vor. Manchmal ist das beim JC-Virus so, wenn nur niedrige Viruslasten vorliegen. Dann kann man den Nachweis nur über eine Hirnbiopsie führen.
Nachweis von SARS-CoV-2 im ZNS
Diesen Grundgedanken nach stellt sich die Frage, kann man eine ZNS-Infektion von SARS-CoV-2 per PCR oder erhöhtem Antikörper-Spezifitätsindex nachweisen? In der Anfangsphase der Pandemie fanden sich in vielen Papern Hinweise auf häufige Nachweise von SARS-CoV-2-RNA im Liquor, so zum Beispiel auch in den oben zitierten von Fotuhi et al. und Yachou et al..
In den Arbeiten von Matschke et al. und Ermis et al. gelang der Nachweis von SARS-CoV-2 per PCR oder Antikörper-Spezifitätsindex maximal in 50% der Fälle, bei Helms et al. hingegen gar nicht. Eine Metaanalyse von Li et al. zeigt wiederum ein etwas anderes Bild. Da es eine Metaanalyse ist und auch noch aus diesem Jahr werde ich die Ergebnisse der Arbeit im Folgenden etwas genauer vorstellen:
Die Autoren fassten die Ergebnisse von 97 Studien mit insgesamt 468 COVID-19-Patienten, die eine Liquoruntersuchung erhalten hatten zusammen. In 25 Studien und bei insgesamt 30 Probanden wurde ein positiver PCR-Nachweis gefunden. Das sind nur 6,4% der Gesamtpopulation. Bei knapp 32% wurde ein entzündliches Liquorsyndrom mit erhöhter Leukozytenzahl im Liquor gefunden, bei knapp 43% eine Erhöhung der Gesamt-IgG im Liquor. Ein Antikörper-Spezifitätsindex wurde bei keinem der PCR-positiven Patienten erhoben.
Bei den 438 Probanden mit negativem PCR-Test im Liquor fand sich in 30% ein entzündliches Liquorsyndrom, bei 46,6% eine Erhöhung des Liquor Gesamt-IgG, bei 46% ein Nachweis von SARS-CoV-2-Antikörpern im Liquor, aber nur bei 23% eine intrathekale Immunglobulinsynthese, das heißt bei 23% stammten die COVID-19-Antikörper aus dem Blut.
Die Prätest-Wahrscheinlichkeit für einen positiven Liquorbefund (PCR oder ASI) scheint extrem von der Klinik der Patienten abzuhängen, die höchsten Nachweis-Wahrscheinlichkeiten bestanden bei Enzephalitiden, schweren Enzephalopathien und Guillain-Barré-Syndromen. Bei fehlender oder unspezifischer neurologischer Symptomatik gelang in der Regel kein SARS-CoV-2-Nachweis im Liquor.
Die Studie untersuchte auch 28 Autopsie-Studien mit insgesamt 202 Patienten, die mit oder an COVID-19 verstorben waren. Hier wurde bei 108 Probanden nach SARS-CoV-2-RNA gesucht und bei 52% gefunden und bei 85 Probanden nach viralen Proteinen (in der Regel Spike-Protein) und bei knapp 30% gefunden. Die meisten Nachweise gelangen aus der Riechschleimhaut (58%) (nun ja, ist mal nicht so ganz das ZNS), dem Riechhirn (26%), dem Großhirn (34%) und dem Hirnstamm (32%). Hier war es so, dass ein positiver Nachweis vor allem dann gelang, wenn „strukturelle Auffälligkeiten“ vorhanden waren, also eine Mikrogliaaktivierung oder Lymphozyteninfiltrate.
Zusammenfassend kann man also sagen, dass in ca. 6% aller untersuchten Fälle ein positiver Liquor-PCR-Test gelang und in 23% aller Fälle ein Nachweis einer intrathekalen spezifischen Immunglobulinsynthese gegen SARS-CoV-2. Das ist nicht nichts, aber im Vergleich zu anderen Erkrankungen des Nervensystems eher auffallend wenig. Für viele Erkrankungen (VZV-Infektionen, Neuroborreliose, usw.) wird ein positiver Liquorbefund als diagnostisches Kriterium gefordert.
Wo man weiterlesen kann
Helms, J., Kremer, S., Merdji, H., Clere-Jehl, R., Schenck, M., Kummerlen, C., Collange, O., Boulay, C., Fafi-Kremer, S., Ohana, M., Anheim, M., & Meziani, F. (2020). Neurologic Features in Severe SARS-CoV-2 Infection. New England Journal of Medicine, 382(23), 2268–2270. https://doi.org/10.1056/nejmc2008597
Li, Y., Zhang, Y., & Tan, B. (2021). What can cerebrospinal fluid testing and brain autopsies tell us about viral neuroinvasion of SARS‐CoV‐2. Journal of Medical Virology, 93(7), 4247–4257. https://doi.org/10.1002/jmv.26943
Matschke, J., Lütgehetmann, M., Hagel, C., Sperhake, J. P., Schröder, A. S., Edler, C., Mushumba, H., Fitzek, A., Allweiss, L., Dandri, M., Dottermusch, M., Heinemann, A., Pfefferle, S., Schwabenland, M., Sumner Magruder, D., Bonn, S., Prinz, M., Gerloff, C., Püschel, K., … Glatzel, M. (2020). Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. The Lancet Neurology, 19(11), 919–929. https://doi.org/10.1016/S1474-4422(20)30308-2
Ermis, U., Rust, M. I., Bungenberg, J., Costa, A., Dreher, M., Balfanz, P., Marx, G., Wiesmann, M., Reetz, K., Tauber, S. C., & Schulz, J. B. (2021). Neurological symptoms in COVID-19: a cross-sectional monocentric study of hospitalized patients. Neurological Research and Practice, 3(1), 17. https://doi.org/10.1186/s42466-021-00116-1
Führt eine Infektion mit SARS-CoV-2 zu bleibenden kognitiven Störungen und erhöht sie eine spätere Entwicklung einer neurodegenerativen Erkrankung? Dieses Thema zieht sich seit dem Spätsommer 2020 durch die wissenschaftliche Diskussion, auch und insbesondere bei der Frage, ob man eigentlich mild verlaufende COVID-Infektionen wie bei Kindern und Jugendlichen nicht einfach zulassen sollte oder ob man das unbedingt verhindern muss.
Klar ist, bei einem relativ neuen Virus kann es keine definitiven Aussagen geben. Es ist vielmehr eine Suche nach Indizien, die sich derzeit auftut. Dabei gibt es verschiedene Hauptargumentation-Linien.
Die Indizien
Neuropsychologische Defizite durch eine SARS-CoV-2-Infektion
Ein Ausgangspunkt der Frage von kognitiven Defiziten ist häufig die Arbeit von Hampshire et al., die ich ja auch schon einmal imLong Covid-Artikel vorgestellt hatte. Hier wurde eine IQ-Abnahme um bis zu sieben Punkte durch eine COVID-Infektion postuliert. An der Studie gibt es durchaus ernstzunehmende methodische Kritik (das kann man ebenfalls im verlinkten Blogbeitrag nachlesen), sie ist nach meiner Wahrnehmung in der Folge auch in der wissenschaftlichen Diskussion etwas in der Versenkung verschwunden.
Eigentlich interessanter, wenn auch vom Patientenumfang deutlich kleiner, ist eine Untersuchung aus dem UKE. Hier wurden 18 Probanden, die eine milde bzw. moderate COVID-Infektion durchgemacht hatten im Mittel 85 Tage nach der Infektion neuropsychologisch getestet, zudem sind umfangreiche laborchemische Tests erfolgt. Verglichen wurde die Kohorte mit 10 gesunden Kontrollprobanden. Die COVID-Erkrankten schnitten erheblich schlechter in einem standardisierten Test für leichte kognitive Störungen ab, als die Kontrollgruppe. Es bestand keine Assoziation zu typischen Long Covid-Symptomen, ebenso wenig zu den umfangreichen untersuchten immunologischen Laborparametern, der gemessenen Viruslast bei der Diagnose der COVID-Erkrankung oder zur Schwere des Krankheitsverlaufes und den erhaltenen Medikamenten. Die Autoren ziehen zunächst den Vergleich zu anderen postinfektiösen Erschöpfungssyndromen, z.B. nach EBV-Infektion oder Influenza (siehe auch hier), stellen dann aber fest:
Our data indicate that neurocognitive deficits after recovery from COVID-19 are independent from fatigue and mood alterations and therefore might be different from the classical post-viral syndrome (Perrin et al., 2020) but a specific post-COVID-19 manifestation.
Der erwähnte Artikel von Perrin et al. ist ein letter to the editors, in dem ein Fallbericht eines Long Covid-Syndroms geschildert wird (Link)
Ich bin mir allerdings nach den Recherchen zum Thema Long Covid nicht so sicher, ob man diese Unterscheidung klinisch sauber machen kann. Nahezu alle Studien zu Long Covid haben bei der Symptomabfrage Mehrfachnennungen erlaubt (was ja auch Sinn macht), aber nirgendwo wird ersichtlich, dass es eine Trennung der Angabe von kognitiven Defiziten mit und ohne Assoziation mit chronischer Erschöpfung gegeben hat. Dazu kommt, dass die Autoren ja betonen, dass die Defizite zum großen Teil subklinisch waren und von den Probanden gar nicht unbedingt bemerkt wurden. Bei den Long Covid-Studien (und auch Untersuchungen zu anderen postinfektiösen Erschöpfungssyndromen) geht es aber in der Regel um subjektiv bemerkte Beschwerden.
Wo man weiterlesen kann
Hampshire, A., Trender, W., Chamberlain, S. R., Jolly, A. E., Grant, J. E., Patrick, F., Mazibuko, N., Williams, S. C., Barnby, J. M., Hellyer, P., & Mehta, M. A. (2021). Cognitive deficits in people who have recovered from COVID-19. EClinicalMedicine, 000, 101044. https://doi.org/10.1016/j.eclinm.2021.101044
Woo, M. S., Malsy, J., Pöttgen, J., Seddiq Zai, S., Ufer, F., Hadjilaou, A., Schmiedel, S., Addo, M. M., Gerloff, C., Heesen, C., Schulze Zur Wiesch, J., & Friese, M. A. (2020). Frequent neurocognitive deficits after recovery from mild COVID-19. Brain Communications, 2(2), 1–9. https://doi.org/10.1093/braincomms/fcaa205
Die PET-Studien
Im Beitrag zur Pathogenese von Long Covid (Link) hatte ich eine der mittlerweile mehreren PET-Studien zum Thema Neuro-COVID schon einmal vorgestellt: Die Arbeit von Hosp et al. und die Anschluss-Studie von Blazhenets et al..
Kurz zusammengefasst wurden 29 schwer betroffene COVID-Patienten im Schnitt einen Monat nach Infektion per FDG-PET untersucht, ebenso eine alterskorrelierte Kontrollgruppe. Herausgefunden wurde ein für neurodegenerative Erkrankungen untypisch verteilter Glukose-Hypometabolismus mit frontaler und parietaler Betonung, einhergehend mit kognitiven – nicht delirtypischen – Defiziten. In einer Folgeuntersuchung nach sechs Monaten waren kognitive Beschwerden und Hypometabolismus teilregredient. Eine kleinere Studie (Morand et al.) mit sieben Kindern, die ein ähnlichen Hypometabolismus zeigten, sorgte für wilde Diskussionen in den sozialen Netzwerken. Größte Schwäche der Kinder-Studie ist die fehlende Kontrollgruppe und die nur bei drei der sieben Kinder bestätigte COVID-Infektion.
Was die Autoren der PET-Studien betonen, ist in der Regel ein COVID-spezifisches Hypometabolismus-Muster und die fehlenden neuropsychologischen Befunde, die auf ein bestehendes Delir hindeuten würden. Dies steht allerdings im diametralen Widerspruch zu einer großen Metaanalyse über COVID-induzierte neuropsychologische und psychiatrische Folgen (Rogers et al.), in der ein Delir bei 27,9% aller COVID-Patienten eines der häufigsten Symptome war.
Wo man weiterlesen kann
Blazhenets, G., Schröter, N., Bormann, T., Thurow, J., Wagner, D., Frings, L., Weiller, C., Meyer, P. T., Dressing, A., & Hosp, J. A. (2021). Slow but evident recovery from neocortical dysfunction and cognitive impairment in a series of chronic COVID-19 patients. Journal of Nuclear Medicine, jnumed.121.262128. https://doi.org/10.2967/jnumed.121.262128
Hosp, J. A., Dressing, A., Blazhenets, G., Bormann, T., Rau, A., Schwabenland, M., Thurow, J., Wagner, D., Waller, C., Niesen, W. D., Frings, L., Urbach, H., Prinz, M., Weiller, C., Schroeter, N., & Meyer, P. T. (2021). Cognitive impairment and altered cerebral glucose metabolism in the subacute stage of COVID-19. Brain, 1–14. https://doi.org/10.1093/brain/awab009
Morand, A., Campion, J. Y., Lepine, A., Bosdure, E., Luciani, L., Cammilleri, S., Chabrol, B., & Guedj, E. (2021). Similar patterns of 18F-FDG brain PET hypometabolism in paediatric and adult patients with long COVID: a paediatric case series. European Journal of Nuclear Medicine and Molecular Imaging, 0123456789. https://doi.org/10.1007/s00259-021-05528-4
Rogers, J. P., Chesney, E., Oliver, D., Pollak, T. A., McGuire, P., Fusar-Poli, P., Zandi, M. S., Lewis, G., & David, A. S. (2020). Psychiatric and neuropsychiatric presentations associated with severe coronavirus infections: a systematic review and meta-analysis with comparison to the COVID-19 pandemic. The Lancet Psychiatry, 7(7), 611–627. https://doi.org/10.1016/S2215-0366(20)30203-0
Die MRT-basierte Studie
Eine der faszinierendsten Arbeiten zu dem Thema ist meines Erachtens ein Paper, was bislang nur als Preprint existiert. Das Konzept der Autoren war, sich der UK Biobank (Link englische Wikipedia) und der darin hinterlegten MRT-Aufnahmen zu bedienen, die vor der COVID-Pandemie angefertigt wurden. 401 Biobank-Teilnehmer mit einer zwischenzeitlich erlittenen COVID-19-Infektion und eine Kontrollgruppe von 384 Probanden wurden nach ein zweites Mal per cMRT untersucht, im Schnitt 3 Jahre nach der Erstuntersuchung. Untersuchungs- und Kontrollgruppe wurden für folgende Confunder angeglichen: Die Vorerkrankungen Blutdruck und Diabetes mellitus, den Body-Mass-Index, den sozioökonomischen Status und für den Sucht- und Genussmittelkonsum Rauchen und regelmäßiger Alkoholkonsum. Die Studienteilnehmer mussten einen neuropsychologischen Test (trail making test) absolvieren.
Die MRT-Aufnahmen vor und nach der COVID-Infektion wurden automatisiert ausgewertet und statistisch aufbereitet. Mit diesen Verfahren (also keiner klassischen Befundung, bei der sich ein Radiologe beide Aufnahmen anschaut und vergleicht) konnten eine Abnahme der Dicke der grauen Substanz im linken orbitofrontalen und beidseitigen parahippocampalen Kortex, sowie im Bereich des Riechhirns bei den SARS-CoV-2 positiven Probanden gezeigt werden. Es schnitten die SARS-CoV-2 positiven Studienteilnehmer deutlich schlechter als die Kontrollgruppe in der neuropsychologischen Testung ab. Je kränker und älter die Probanden waren, desto deutlicher war der Effekt. Eine Erholung im engeren Sinne ließ sich nicht beobachten, eine länger zurückliegende Infektion hatte keinen Einfluss auf die Untersuchungsergebnisse.
Die Autoren durchdenken zwei mögliche Erklärungsmodelle für ihre Beobachtungen: Eine neuronale Degeneration, ausgehend vom Riechhirn (bei dem eine SARS-CoV-2-Infektion auf Grund der häufigen Hyposmie gemeinhin angenommen wird). Durch die verschiedenen Faserverbindungen, die vom Riechhirn ausgehen, müsste nicht mal eine weitergehende ZNS-Infektion stattfinden. Ein ähnliches Degenerationsmuster wurde für Influenza-Viren in der Vergangenheit schon gezeigt. Die zweite Hypothese ist eine ausgeprägte (auto)inflammatorische Reaktion (der berühmte Zytokinsturm).
Wo man weiterlesen kann
Douaud, G., Lee, S., Alfaro-Almagro, F., Arthofer, C., Wang, C., Lange, F., Andersson, J. L. R., Griffanti, L., Duff, E., Jbabdi, S., Taschler, B., Winkler, A., Nichols, T. E., Collins, R., Matthews, P. M., Allen, N., Miller, K. L., & Smith, S. M. (2021). Brain imaging before and after COVID-19 in UK Biobank. MedRxiv : The Preprint Server for Health Sciences. https://doi.org/10.1101/2021.06.11.21258690
Spike-Protein und ß-Amyloid: Theoretische Betrachtungen
Wie es manchmal so ist, wurde immer wieder eine sehr theoretische und aus der Grundlagenforschung stammende Arbeit zu dem Thema zitiert, die man aber einmal einordnen muss, da sie sonst vor allem Fragen hinterlässt. In der Kurzversion des Blogbeitrages (Link) hatte ich schon mal etwas dazu geschrieben.
Die Arbeit von Idrees und Kumar hat mit einer Computersimulation mit dem HDOCK server (einer webbasierten Lösung, mit der Proteininteraktionen simuliert werden können, Link pubmed) mögliche Interaktionen und Bindungen zwischen dem Spike-Protein von SARS-CoV-2 und den vier Proteinen, welche wir von den neurodegenerativen Erkrankungen kennen (ß-Amyloid, tau-Protein, a-Synuclein und TDP-43, hier kann man zu dem Thema neuropathogene Proteine etwas weiterlesen) ermittelt. Es handelt sich also um ein theoretisches Computer-Experiment. Heraus kam, dass das Spike-Protein durchaus mit den neurodegenerativen Proteinen interagieren und auch an diese mit einer erhöhten Affinität binden kann. Die Autoren verweisen auf eine andere Arbeit, die die Induktion von neurodegenerativen Erkrankungen durch verschiedene Virusinfektionen nahegelegt habe (Zhou et al., zu der ich weiter unten noch was schreibe) und schlussfolgern, dass es sich mit SARS-CoV-2 ähnlich verhalten könnte.
Wo man weiterlesen kann
Idrees, D., & Kumar, V. (2021). SARS-CoV-2 spike protein interactions with amyloidogenic proteins: Potential clues to neurodegeneration. Biochemical and Biophysical Research Communications, 554, 94–98. https://doi.org/10.1016/j.bbrc.2021.03.100
ACE und Neuro-COVID
Im Long Covid-Blogthema ging es schon einmal um das die Bindung des Spike-Proteins an ACE-Rezeptoren (hier kann man dazu weiterlesen, Link), aber wenn man das Thema Neurodegeneration und Covid verstehen möchte, muss man sich wohl oder übel etwas näher damit beschäftigen. Sehr empfehlenswert sind dazu die Review-Paper von Miners et al. und von Pacheco-Herrero et al.. Beide Autorengruppen geben einen umfassenden Überblick über die angenommenen Mechanismen zur möglichen Neurodegeneration verstärkenden von SARS-CoV-2. Das Thema ist sehr theoretisch und grundlagenforschungslastig, aber ich versuch mal mein bestes:
Der Schlüssel für die Invasion von Körperzellen durch SARS-CoV-2 ist die Bindung des Virus an das Angiotensin-konvertierende Enzym-2 (ACE-2). ACE-2 wird auf vielen Neuronen exprimiert, auch und insbesondere im Riechhirn, im Hippocampus und generell im Temporallappen, zudem auf vielen Gliazellen. Aus Tierexperimenten weiß man zudem, dass das SARS-1-Virus den Nervus vagus infiltrieren und hier retrograd in den Hirnstamm gelangen kann. Dieser retrograde axonale Transport ist ja ein Phänomen, welches sehr viele Viren beherrschen (nicht nur Tollwut-Viren und Viren der Herpesgruppe) und mit dem sich viele Viren durch den Körper „bewegen“. Es ist naheliegend, dass wenn SARS-1-Virus das kann, SARS-CoV-2 derartige Features auch beherrscht, v.a. weil in Autopsie-Studien vereinzelt SARS-CoV-2 im Hirnstamm und häufig (z.B. Matschke et al.) dort eine lymphozytäre Infiltration und Mikrogliaaktivierung nachgewiesen wurde, die ja in der Regel eine Reaktion auf irgendetwas andere ist, z.B. eine massive Immunreaktion.
Einen guten Hinweis in diese Richtung gibt die Arbeit von Heneka et al.. Die Autoren beschreiben die vielen verschiedenen Zytokine, die v.a. bei schweren COVID-19-Verläufen nachgewiesen wurden, namentlich: Interleukin-1β, Interleukin-2, Interleukin-2-Rezeptor, Interleukin-4, Interleukin-10, Interleukin-18, Interferon-γ, C-reaktives Protein, Granulozytenkolonie-stimulierender Faktor, Interferon-γ, CXCL10, monocyte chemoattractant protein 1, macrophage inflammatory protein 1-α und Tumornekrosefaktor-α bei parallel aber abfallenden T-Zell-Spiegeln im Blut, welche man aus der häufigen Leukopenie ableiten könne und die Aktivierung von Inflammasomen, (Link Wikipedia), also von Proteinkomplexen, die Entzündungsreaktionen auslösen können. Besonders der Protein-Komplex mit dem klangvollen Namen NLRP3 wurde auch bei anderen schweren Erkrankungen (v.a. bei Sepsis-Patienten, aber auch bei Influenza-Infektionen) als ein wesentlicher Bestandteil in der Pathogenese identifiziert und ist in Laborexperimenten mir einer Induktion von Peptid-Ablagerungen wie Amyloid-ß assoziiert gewesen. Die Autoren leiten daraus eine mögliche Induktion von Demenzerkankungen durch SARS-CoV-2 ab, beziehen sich aber klinisch v.a. auf die Assoziation von NLRP3 bei der Sepsis mit Neurodegeneration.
Mit der Infektion mit SARS-CoV-2 scheint es zu einer Spaltung von ACE-Rezeptoren und einer Internalisierung dieser zu kommen, wodurch dem Körper weniger ACE-Rezeptoren zur Verfügung stehen. Dies führt zu Ungleichgewichten im Renin-Angiotensin-System (Link Wikipedia), welches durch ACE je wesentlich gesteuert wird. Der Theorie nach führt das zu Endothelschäden und einer Dysfunktion der kleinen Gefäße, wenn man so will zu einer Mikroangiopathie. Das ist der Punkt der gemeint ist, wenn gesagt wird: COVID-19 ist eine Erkrankung des Endothels.
Aus der Grundlagenforschung zur Alzheimer-Demenz weiß man wiederum, dass auch hier in der Frühphase der Erkrankung die kleinen Gefäße eine wichtige Rolle zu spielen scheinen und eine Dysfunktion hier zu einer erhöhten Konzentration von neurotoxischen Amyloid-ß-Ansammlungen führt. Dies wiederum bedingt eine Fehlfunktion der Perizyten, also der den Kapillaren anliegenden Bindegewebezellen. Hierdurch wird die Blut-Hirn-Schranke durchlässiger. Das alles führt zu oxidativem Stress mit vermehrter NO-Ausschüttung, was wiederum Mitochondrien-Schäden verursachen kann und zudem eine Hyperphosphorilierung von Tau-Protein induziert. Das hier eine Verbindung zwischen Alzheimer-Pathomechanismen und COVID-19 besteht, wird mit Erkenntnissen aus Tiermodellen unterfüttert, bei denen transgene Mäuse nach Atemwegsinfektionen vermehrte T-Zell-Infiltrationen im Gehirn und eine vermehrte Amyloid-ß-Ablagerung bekamen. Mit zunehmendem Alter und männlichem Geschlecht gibt es weniger ACE-Exprimierung, was der Theorie nach die schwereren Krankheitsverläufe bei COVID-19 und das schlechtere Outcome erklären könnte, so die Autoren. Zudem merken sie an, dass die weiteren Risikofaktoren für ein schlechtes COVID-Outcome Bluthochdruck, Diabetes und Adipositas ebenfalls mit einer Fehlregulation des Renin-Angiotensin-Systems einhergehen. Für die Nicht-Neurologen, vor allem die Intensivmediziner sei erwähnt, dass eine Renin-Angiotensin-System-Dysfuntion auch mit einem höheren Risiko eines ARDS (Link Wikipedia) einhergeht.
Wo man weiterlesen kann
Heneka, M. T., Golenbock, D., Latz, E., Morgan, D., & Brown, R. (2020). Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimer’s Research & Therapy, 12(1), 69. https://doi.org/10.1186/s13195-020-00640-3
Matschke, J., Lütgehetmann, M., Hagel, C., Sperhake, J. P., Schröder, A. S., Edler, C., Mushumba, H., Fitzek, A., Allweiss, L., Dandri, M., Dottermusch, M., Heinemann, A., Pfefferle, S., Schwabenland, M., Sumner Magruder, D., Bonn, S., Prinz, M., Gerloff, C., Püschel, K., … Glatzel, M. (2020). Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. The Lancet Neurology, 19(11), 919–929. https://doi.org/10.1016/S1474-4422(20)30308-2
Miners, S., Kehoe, P. G., & Love, S. (2020). Cognitive impact of COVID-19: looking beyond the short term. Alzheimer’s Research & Therapy, 12(1), 170. https://doi.org/10.1186/s13195-020-00744-w
Pacheco-Herrero, M., Soto-Rojas, L. O., Harrington, C. R., Flores-Martinez, Y. M., Villegas-Rojas, M. M., León-Aguilar, A. M., Martínez-Gómez, P. A., Campa-Córdoba, B. B., Apátiga-Pérez, R., Corniel-Taveras, C. N., Dominguez-García, J. de J., Blanco-Alvarez, V. M., & Luna-Muñoz, J. (2021). Elucidating the Neuropathologic Mechanisms of SARS-CoV-2 Infection. Frontiers in Neurology, 12(March 2020), 1–19. https://doi.org/10.3389/fneur.2021.660087
Was bedeutet das nun?
Allgemeines Prinzip oder SARS-CoV-2-Besonderheit?
Der Aspekt, der mich am meisten an dem Thema interessiert ist, ob es sich bei den Erkenntnissen um SARS-CoV-2-spezifische Prozesse handelt oder am Ende um allgemeingültige Pathomechanismen, die wir bei anderen Infektionskrankheiten auch sehen können (vielleicht in der Vergangenheit aber nicht genau genug hingeschaut haben). Dieser Gedanke ist in so fern naheliegend, da zu keinem Zeitpunkt in der Geschichte der medizinischen Forschung mit vergleichbarem personellen und finanziellen Aufwand Forschung zu einem einzigen Krankheitsbild betrieben wurde.
Was beim Lesen der Studien und Review-Paper auffällt, dass nahezu überall andere virale Infektionskrankheiten als Referenz und Vergleich genannt werden, interessanterweise meistens Influenza, RS-Virus und die Viren der Herpesgruppe. Das betrifft die Mechanismen der Neuroinvasion über das Riechhirn, Atrophien von Nervenzellschichten nach Infektion und postinfektiös auftretenden kognitiven Störungen. Dass nach anderen Infektionen Gedächtnisstörungen auftreten, ist kein großes Geheimnis, insbesondere nicht, wenn ein komorbides Delir vorliegt. Nicht ohne Grund werden standardisierte neuropsychologische Testungen mit der Frage nach einer Demenzerkrankung in der Regel frühestens sechs Monate nach einem Delir durchgeführt.
Der Infektionsmechanismus über die ACE-2-Infiltration scheint hingegen ziemlich Corona-Virus-spezifisch zu sein, allerdings nur für SARS-CoV-2 und für das SARS-1-Virus und nicht für MERS und verschiedene Tier-Coronaviren (vgl. Ng Kee Kwong et al.)
Was man zum Zusammenhang von Virusinfektionen und Neurodegeneration weiß
In mehreren Papern wird auf die Arbeit von Zhou et al. zum Thema Neurodegeneration und Viruserkrankungen verwiesen. Auch hierzu eine kurze Zusammenfassung: Es handelt sich um eine relativ lange Review-Arbeit, in der für verschiedene (neurotrope) Viren die Möglichkeit einer Assoziation mit neurodegenerativen Erkrankungen erläutert werden. Dabei muss man beachten, dass die Arbeit von 2013 ist und einige hier präferierte Mechanismen in den letzten acht Jahren kaum noch diskutiert wurden. Die Autoren betonen eine Assoziation zwischen Herpes-Enzephalitiden und hierdurch wahrscheinlicher auftretenden Demenzerkankungen, ziehen eine (sattsam bekannte) Verbindung zwischen Infektionen mit Viren aus der Herpes-Gruppe und der Entwicklung einer Multiplen Sklerose und zwischen Influenza-Infektionen und der Entwicklung von Parkinson. Es wird auch der historische Vergleich bemüht, dass nach verschiedenen Grippe-Pandemien verstärkt postinfektiöse Parkinson-Erkrankungen beobachtet wurden.
Einschränkend muss erwähnt werden, dass man hier zwar statistische Korrelationen zeigen kann, dass bis heute aber die Virus-Hypothese bei der Multiplen Sklerose nicht bewiesen werden konnte (anders als der Einfluss des Vitamin-D-Spiegels), ebenso wenig Influenza-Infektionen als Auslöser von Parkinson. Als statistisch evidente Risikofaktoren für die Entwicklung von Parkinson-Erkrankungen gelten weiterhin fehlender Koffein- Alkohol- und Nikotinkonsum, Kopftraumata, Obstipationsneigung, depressive Störungen, Angsterkrankungen, Beta-Blocker-Einnahme, kein Bluthochdruck, Arbeiten in der Landwirtschaft, Leben auf dem Land und Pestizid-Exposition (vgl. Pan-Montojo und Reichmann und Lill und Klein).
Man muss sogar sagen, dass in den letzten Jahren das Verständnis der Entstehung neurodegenerativer Erkrankungen riesige Fortschritte gemacht hat (insbesondere was die Prion-artige Ausbreitung der pathogenen fehlgefalteten Proteine betrifft), hinsichtlich des Themas einer ggfs. Virus-bedingten Triggerung hingegen so gut wie nichts getan hat.
Wo man weiterlesen kann
Ng Kee Kwong, K. C., Mehta, P. R., Shukla, G., & Mehta, A. R. (2020). COVID-19, SARS and MERS: A neurological perspective. Journal of Clinical Neuroscience, 77(January), 13–16. https://doi.org/10.1016/j.jocn.2020.04.124
Pan-Montojo, F., & Reichmann, H. (2015). Ursache der Parkinson-Krankheit: Braak revisited. Aktuelle Neurologie, 41(10), 573–578. https://doi.org/10.1055/s-0034-1387475
Das ziehe ich in Teil 3. Ich möchte das mit Absicht von der Schilderung der wissenschaftlichen Literatur abgrenzen, da ich denke, dass man die Frage, ob SARS-CoV-2 nun neurodegenerative Prozesse wahrscheinlicher machen kann durchaus unterschiedlich interpretieren kann.
Heute soll es relativ kurz und außer der Reihe um das Thema Führt eine COVID-19-Erkrankung zu einer höheren Wahrscheinlichkeit einer späteren neurodegenerativen Erkrankung? gehen. Anlass war diese Replik hier:
Das darin verlinkte Paper von Idrees und Kumar kannte ich bislang noch nicht, wohl aber ein ähnlich thematisch positioniertes von Pacheco-Herrero et al. (okay, zugegebenermaßen weil es im DGN-Covid-Paper-Journal-Club besprochen wurde (Link).
Kurze Zusammenfassung
SARS-CoV-2 spike protein interactions with amyloidogenic proteins: Potential clues to neurodegeneration
Diese Arbeit ist eine Arbeit aus der Grundlagenforschung. Es wurden mit einer Computersimulation mit dem HDOCK server (einer webbasierten Lösung, mit der Proteininteraktionen simuliert werden können) mögliche Interaktionen und Bindungen zwischen dem Spike-Protein von SARS-CoV-2 und den vier Proteinen, welche wir von den neurodegenerativen Erkrankungen kennen (ß-Amyloid, tau-Protein, a-Synuclein und TDP-43, hier kann man dazu etwas weiterlesen) ermittelt. Es handelt sich also um ein theoretisches Computer-Experiment. Heraus kam, dass das Spike-Protein durchaus mit den neurodegenerativen Proteinen interagieren und auch an diese mit einer gewissen Affinität binden kann. Die Autoren verweisen auf eine andere Arbeit, die die Induktion von neurodegenerativen Erkrankungen durch verschiedene Virusinfektionen gezeigt habe (Zhou et al.) und schlussfolgern, dass es sich mit SARS-CoV-2 ähnlich verhalten könnte.
Elucidating the Neuropathologic Mechanisms of SARS-CoV-2 Infection
In dieser Arbeit – welche auch im Mai 2021 erschien – ging es primär um verschiedene Infektionswege von SARS-CoV-2 und eine etwaige Neurotropie. Es ist ein sehr aufwändig gestaltetes Review-Paper, ebenfalls aus der Grundlagenforschung. Die Autoren fassen verschiedene molekularbiologische Aspekte einer SARS-CoV-2-Infektion über ACE-Rezeptoren zusammen (hier kann man dazu weiterlesen Link). Durch die ACE-Aktivierung wird NO ausgeschüttet, welches v.a. Mitochondrien-Schäden verursachen kann, zudem eine Hyperphosphorilierung von Tau-Protein induziert. Auch die Autoren verweisen auf andere Arbeiten, welche eine Assoziation zwischen Virusinfektionen und neurodegenerativen Erkrankungen zeigten.
Wie will man diese Erkenntnisse deuten?
Das ist die große Frage. Verwiesen wird in beiden Arbeiten auf andere Studien, zum Beispiel die von Zhou et al. zum Thema Neurodegeneration und Viruserkrankungen.
Noch ein Paper: Viruses and neurodegeneration
Auch hierzu eine kurze Zusammenfassung. Es handelt sich um eine relativ lange Review-Arbeit, in der für verschiedene (neurotrope) Viren die Möglichkeit einer Assoziation mit neurodegenerativen Erkrankungen erläutert werden. Dabei muss man beachten, dass die Arbeit von 2013 ist und einige hier präferierte Mechanismen in den letzten acht Jahren kaum noch diskutiert wurden. Die Autoren betonen eine Assoziation zwischen Herpes-Enzephalitiden und hierdurch wahrscheinlicher auftretenden Demenzerkrankungen, ziehen eine (sattsam bekannte) Verbindung zwischen Infektionen mit Viren aus der Herpes-Gruppe und der Entwicklung einer Multiplen Sklerose und zwischen Influenza-Infektionen und der Entwicklung von Parkinson. Es wird auch der historische Vergleich bemüht, dass nach verschiedenen Grippe-Pandemien verstärkt postinfektiöse Parkinson-Erkrankungen beobachtet wurden.
Einschränkend muss erwähnt werden, dass man hier zwar statistische Korrelationen zeigen kann, dass bis heute aber die Virus-Hypothese bei der Multiplen Sklerose nicht bewiesen werden konnte (anders als der Einfluss des Vitamin-D-Spiegels), ebenso wenig Influenza-Infektionen als Auslöser von Parkinson. Als statistisch evidente Risikofaktoren für die Entwicklung von Parkinson-Erkrankungen gelten weiterhin fehlender Koffein- Alkohol- und Nikotinkonsum, Kopftraumata, Obstipationsneigung, depressive Störungen, Angsterkrankungen, Beta-Blocker-Einnahme, kein Bluthochdruck, Arbeiten in der Landwirtschaft, Leben auf dem Land und Pestizid-Exposition (vgl. Pan-Montojo und Reichmann und Lill und Klein).
Beunruhigen mich die Paper?
Eher nicht. Und zwar in erster Linie deshalb, weil diese Frage nach der Rolle von Viren in der Genese sowohl von autoimmun-entzündlichen Erkrankungen als auch bei der Entstehung von neurodegenerativen Syndromen seit Jahren (im Endeffekt seit ich Neurologie mache) in ihrer Beantwortung stagniert und man sagen kann: Ja, es ist möglich, aber nicht unbedingt wahrscheinlich.
Idrees, D., & Kumar, V. (2021). SARS-CoV-2 spike protein interactions with amyloidogenic proteins: Potential clues to neurodegeneration. Biochemical and Biophysical Research Communications, 554, 94–98. https://doi.org/10.1016/j.bbrc.2021.03.100
Pacheco-Herrero, M., Soto-Rojas, L. O., Harrington, C. R., Flores-Martinez, Y. M., Villegas-Rojas, M. M., León-Aguilar, A. M., Martínez-Gómez, P. A., Campa-Córdoba, B. B., Apátiga-Pérez, R., Corniel-Taveras, C. N., Dominguez-García, J. de J., Blanco-Alvarez, V. M., & Luna-Muñoz, J. (2021). Elucidating the Neuropathologic Mechanisms of SARS-CoV-2 Infection. Frontiers in Neurology, 12(March 2020), 1–19. https://doi.org/10.3389/fneur.2021.660087
Die Veröffentlichung des aktuellsten Thesenpapiers der Autorengruppe u.a. um Matthias Schrappe, Wissenschaftler der Universität Bremen und Klaus Püschel (ex Institut für Rechtsmedizin des UKE) (Link) führt bei Twitter zu hohen Wellen und zum Trending des Hashtags „DIVIGate“ (Link), allerdings sind 99,9% der Beiträge einfach nur dumm und hanebüchen. Diskutiert wird (mal wieder) nicht.
Auch wenn das Thesenpapier sicherlich mehrere wunde Punkte aufzeigt, hakt es doch m.E. an verschiedenen Stellen. Daher schreibe ich mal eine kurze Besprechung/Erwiderung, etwas subjektiver und mit weniger Quellen als sonst.
Update vom 18.05.:
Die Wellen schlagen weiter hoch, nun hat die DIVI (das heißt übrigens Deutsche Interdisziplinäre Vereinigung für Intensiv- und Notfallmedizin) eine scharf formulierte Stellungnahme veröffentlicht: Link und auch auf Twitter hat sich die Gegenseite in Stellung gebracht. Verschiedene objektivierbare Fehlannahmen in dem Thesenpapier wurden identifiziert und benannt. Nur gesprochen und diskutiert wird weiterhin nicht miteinander, sondern man beschimpft sich gegenseitig und wertet sich ab, auch bei sonst gut recherchierten Serien-Posts:
Schon jetzt ist mehr Mühe und Sorgfalt ins Debunking geflossen als die Autoren selbst für ihr Pamphlet aufgewendet haben. Wir haben alle eigentlich besseres zu tun. Ich selbst hatte mich heute morgen dagegen entschieden, dem hinterher zu recherchieren.
Der "Skandal" liegt also offenbar nicht in einer Manipulation durch die @DIVI_eV, die Herr Schrappe raunend in den Raum stellt, sondern in seiner Unfähigkeit, die Daten richtig zu verstehen.
Ich hangel mich hier weiterhin an der Struktur des Thesenpapiers entlang, ergänze den Beitrag von gestern aber an verschiedenen Stellen mit den neuen Informationen.
Kapitel 1: Akutstationäre Versorgung
Der Beginn des Thesenpapiers liest sich wie eine Fortsetzung der Bertelsmann-Studie zur Krankenstruktur in Deutschland (Link), die seinerzeit ja sehr kontrovers diskutiert wurde. Die Autoren des Thesenpapiers schildern die Schwierigkeiten der Krankenhausplanung in Deutschland und die Bedeutung für Krankenhausträger, im jeweiligen Bettenplan aufzutauchen. Kurz wird die Umstrukturierung der Krankenhauslandschaft in Deutschland aufgegriffen, mit einer Abnahme von Krankenhäusern in kommunaler und freigemeinnütziger Trägerschaft und einer Zunahme von privatwirtschaftlich betriebenen Häusern. Hierüber und über die Tatsache, dass Deutschland die höchste Dichte an Krankenhausbetten in der EU pro 1000 Einwohner hat, kommt zu dem Schluss, dass in Deutschland eine Überversorgung mit stationären Betten besteht, Es wird festgestellt:
„Patienten werden wegen ambulant behandelbarer Erkrankungen häufiger im Krankenhaus aufgenommen als in den meisten anderen EU-Ländern“.
Die genaue Herleitung dieser Feststellung erschließt sich auch bei mehrmaligem Lesen nicht. Im Verlauf drängt sich ein bisschen der Eindruck auf, die Autoren benötigen diese Feststellung als Unterbau für ihre weitere Argumentationskette.
Kapitel 2: Ausstattung mit Intensivbetten im internationalen Vergleich
Zu Beginn des Kapitels wird kurz die sehr heterogene Bevölkerungsstruktur in der EU beschrieben mit sehr vielen hochaltrigen Bewohnern zum Beispiel in Italien und deutlich weniger in Irland. Dann erfolgt eine Einordnung der Intensivbetten pro 100.000 Einwohner im internationalen Vergleich, es wird festgestellt, dass Deutschland mit 33,9 Intensivbetten/100.000 Einwohner einen Spitzenplatz belegt, gerade gegenüber Großbritannien, Spanien und Italien. Hier gibt es wenig kontroverses.
Kapitel 3: Corona-Pandemie im europäischen Ausland: Erste Reaktionen im Frühjahr 2020
Hier erfolgt eine Beschreibung der Maßnahmen, die Länder mit weniger Intensivbetten zu Beginn der COVID-19-Pandemie eingeleitet haben, zum Beispiel durch die Umwidmung „normaler“ Krankenhausbetten in Intensivbetten oder durch die Errichtung von Behelfskrankenhäusern.
Kapitel 4: Verlauf und Vorgehen in Deutschland
Die Autoren beschreiben hier zunächst die in den bisherigen drei Krankheitswellen unterschiedlich betroffenen Patientengruppen. Hier scheint es einen Streit um die korrekte Anzahl der betroffenen Patienten zu geben:
Fehler 1, ziemlich banal und peinlich: Spitzenlast an Covid-Patienten einfach mal mit Gesamtzahl an Covid-Patienten auf ICU gleichgesetzt. Entdeckt von @janoschdahmenhttps://t.co/kpRkHpzUAv
Dies ist nach meinem Verständnis für die Kernaussage des Thesenpapiers aber ziemlich nebensächlich.
Dann wenden die Autoren ihren Fokus auf die gesundheitspolitischen Maßnahmen, insbesondere die Ausgleichszahlungen („Freihaltepauschale“) für ungenutzte Intensivbetten, aber auch die sonstigen Ausgleichszahlungen. Kurz wird auch der starke Rückgang der Non-COVID-Patientenzahlen in der ersten Welle angeschnitten.
Dann wird es interessant, die Autoren schreiben zwar
Neben diesen Maßnahmen darf man jedoch nicht die gesellschaftliche und institutionelle Stimmungslage aus dem Auge verlieren. Ohne Zweifel führte und führt die CoViD-19- Pandemie zu einer extremen Belastung des Gesundheitswesens und der Krankenhäuser.
und fassen auch die jeweiligen Schwierigkeiten in den Krankenhäusern in der ersten Welle (fehlende Schutzausrüstungen, allgemeine Unwissenheit) und der zweiten Welle (Ausbrüche beim Personal, hoher Krankenstand, viele hochaltrige, schwer betroffene Patienten) zusammen, kommen aber zum Schluss, dass in der dritten Welle die Sache sich entspannt hat. Und dann geht es gleich weiter mit noch einer Runde Ausgleichszahlungen und Prämien für neue Intensivbetten. Das ist m.E. der erste Punkt, an dem man Kritik äußern muss:
Die Autoren lassen den durch COVID-19 gerade auf den Intensivstationen veränderten Alltag außer acht. Es ist nämlich nicht damit getan, dass alle genug FFP2-Masken und Schutzkittel haben und Pauschalen geflossen sind, sondern es macht einen erheblichen Unterschied für viele Beschäftigte im Gesundheitswesen und hier gerade in der Intensivmedizin aus, ob man von Zimmer zu Zimmer gehen, zwischendurch was trinken kann und auch mal kurz mit den Kollegen klönt, oder ob man – wie auf einer COVID-Intensivstation – teilweise stundenlang in voller Schutzmontur in einem Patientenzimmer feststeht. Gerade weil die Pflege von COVID-Intensivpatienten sehr aufwändig ist und gerade wenn z.B. Bauchlagerung oder eine ECMO erforderlich sind. Ein wenig Einblick, wie sich „das anfühlt“, gibt die COVID-19 Reportage aus der Charité (Link). Hier kann man natürlich einwenden, dass es interessant wäre ein Vorher-Nachher-Vergleich zu machen, also ob sich die Situation auf den Intensivstationen durch COVID-19 wirklich so gewandelt hat und ob die allgemeine Erschöpfung und der ewig gleiche Trott nicht vorher auch genauso da waren. Das mag sicherlich so sein, aber als Katalysator hat COVID-19 auf jeden Fall gewirkt. Was auch außer acht gelassen wird, dass im 16. Monat der Pandemie in Deutschland der „Akku“ bei vielen Beschäftigten im Gesundheitswesen einfach erschöpft ist und was hierfür Gründe sein könnten. Ich würde behaupten, das liegt ganz wesentlich an allem, was derzeit wegfällt. Das Miteinander mit den Kollegen, der Schnack, der Kaffee und ggfs. die gemeinsame Zigarette zwischendurch und zwar ohne Maske und ohne Abstand. Das Angehörigen-Ersatz-Sein, durch die Besuchsverbote, die Schwierigkeiten Therapieentscheidungen mit Angehörigen telefonisch und nicht vor Ort am Patientenbett zu besprechen. Die Abstreiche-Logistik vor Verlegung in Pflegeheime, Rehakliniken usw.
Kapitel 5: Entscheidendes Defizit: unzureichende Datenlage zur Nutzung
Das Kapitel, in dem die Autoren sicherlich ihren Stich machen. Das Kapitel beginnt mit der Problematik der Drohkulisse hinsichtlich Triage, Überlastung von Intensivmedizin und immer jüngeren Intensivpatienten, wie sie verwendet wurde, um schärfere Corona-Maßnahmen durchzusetzen. Hier gab es ja eine Menge öffentlichwirksam vorgetragener steiler Thesen
(1) Die Visualisierung ist gut und zeigt, dass im Vergleich zu Beginn 2. Welle die 3. Welle mit deutlich mehr Kindern losgeht. Zwar weniger sehr Alte, aber viele in Gruppe 40-70 J. Durchschnittsalter auf Intensivstation ist jetzt nur noch ca 58 J, diese Patienten liegen lange. https://t.co/Wgo59gRDOr
— Prof. Karl Lauterbach (@Karl_Lauterbach) March 24, 2021
(3) Jeder der das für „Panikmache“ hält, sollte sich auf Covid Intensivstationen einmal umsehen: 40-50 Jährige Eltern sind dort keine Rarität. Daher müssen wir als Gesellschaft die nächsten Wochen noch einmal Lockdown machen. Ab Ende Mai sinkt wegen der Impfung die Fallzahl stark
— Prof. Karl Lauterbach (@Karl_Lauterbach) April 19, 2021
Sein Vorwurf, es sei Angst geschürt worden, verkennt die Situation des Frühjahrs 2020. Tatsächlich herrschte im März des vergangenen Jahres Angst davor, dass zahlreiche Patientinnen und Patienten nicht mehr ausreichend versorgt, insbesondere beatmet, werden könnten. Die Sorge war angesichts der Situation in Italien, Frankreich und vielen anderen Ländern begründet.
So ganz von der Hand zu weisen ist der Punkt aber nicht, siehe die Äußerungen von Karl Lauterbach.
Hauptpunkt des Thesenpapier ist aber das – kongruent zu anderen COVID-19-Themen (vergleiche z.B. hier zum Thema Fallzahlenvorhersage) – extrem schlechte Monitoring von so ziemlich allem in der Pandemie in Deutschland, hier halt das der Intensivbettenbelegung (Fehlende Angaben zum Alter, Geschlecht, Komorbiditäten, Grund der Aufnahme auf die Intensivstation (COVID oder ein anderer Grund, aber ein positiver COVID-Befund). Die Schlussfolgerung
Analog zum gängig gewordenen Ausdruck „Mortalität mit oder an Corona“ wäre hier „Intensivpflichtigkeit mit oder wegen Corona“ die adäquate Umschreibung. Genaue Daten zu dieser Differenzierung liegen nicht vor. Wenn man von einer Prävalenz in der Bevölkerung von 1% ausgeht, wären dies bei rund 20.000 Intensivpatienten immerhin 200 Patienten, die gar nicht wegen CoViD-19 intensivpflichtig werden (jedoch dann als solche gepflegt und behandelt werden müssen). Jüngere Patienten mit schweren Grunderkrankungen wären hiervon stärker betroffen als es für ältere Patienten der Fall ist, denn bei Jüngeren ist die Chance, allein wegen SARS-2/CoViD-19 intensivpflichtig zu werden, sehr gering.
kann man sicherlich unterschreiben. Die öffentlich kommunizierte erhöhte Krankenhausbehandlungsbedürftigkeit jüngerer Patienten schnurrt dann in sich zusammen, wenn die Autoren darlegen können, dass sich die absolute Zahl unter 80-jähriger Intensiv-Corona-Patienten zwar erhöht hat, nicht aber der prozentuale Anteil in Bezug auf die Alterskohorten. Übersetzt heißt das: Weil die meisten hochaltrigen Menschen geimpft sind, nimmt die Zahl über 80-jähriger COVID-Patienten ab, es werden jetzt halt überwiegend unter 80-jährige behandelt. Bei hohen Fallzahlen resultieren hohe absolute Zahlen, eine prozentuale Zunahme schwer betroffener jüngerer COVID-Patienten lässt sich aber nicht beobachten.
Kapitel 6: Wertung aufgrund internationaler Daten
Hauptpunkt in diesem Kapitel ist die Tatsache, dass Deutschland in Bezug auf gemeldete COVID-Fälle und Intensivbetten im internationalen Vergleich sehr komfortabel aufgestellt ist, dass aber der Anteil an auf Intensivstationen behandelter COVID-Patienten in Deutschland deutlich höher ist, als in anderen Ländern.
In Deutschland wurden am 27.4.2021 61% der hospitalisierten CoViD-19-Patienten auf Intensivstationen behandelt, während dies nur für 25% der Patienten in der Schweiz oder 11% der Patienten in Italien zutraf.
Hierfür gibt es zunächst keine schlüssige Erklärung. Postuliert wird im Thesenpapier eine „Überversorgung“, was im Angesicht der Triage-Diskussion tatsächlich skurril wäre. Nun hat gerade die in dem Thesenpapier gescholtene DIVI aber eine sehr gute und kritische Arbeit zum Thema Überversorgung veröffentlicht (Link).
Im Zuge der Erwiderungen auf das Thesenpapier finden sich z.B. folgende Gegenrechnung:
Im Papier von Prof. #Schrappe scheint es noch einen weiteren wichtigen Fehler zu geben, und zwar bezüglich der Aussage, dass in Deutschland angeblich 44 % der Covid-Patienten, die im Krankenhaus liegen, auf Intensivstationen behandelt werden. (1/10)
Das berechnet er in einer Tabelle aus der angeblichen Zahl der hospitalisierten Covid-19-Patienten und der Zahl der Covid-19-Patienten auf Intensivstationen. (2/10) pic.twitter.com/AXoPHHDS2V
Das Problem: Während die Corona-Intensivpatienten im DIVI-Intensivregister täglich exakt erfasst werden, gibt es meines Wissens keine verlässliche Zahl zur jeweils aktuellen Zahl der Corona-Patienten, die im Krankenhaus liegen. (3/10)
Die Zahl aus dem RKI-Bericht, mit der Herr Schrappe arbeitet, sagt jedenfalls nur aus, wie viele Menschen innerhalb einer Woche aufgrund von Corona ins Krankenhaus mussten. Um zu wissen, wie viele dort gleichzeitig liegen, braucht man die durchschnittliche Liegedauer. (4/10)
Wenn man mit diesem Wert die 8112 Krankenhaus-Fälle in der Tabelle, die sich auf eine Woche beziehen, umrechnet, bekommt man 11.589 Corona-Patienten, die sich zu dem Zeitpunkt gleichzeitig im Krankenhaus befanden. (6/10)
Und damit sinkt der angeblich extrem hohe Anteil der Corona-Intensivpatienten an den Corona-Krankenhauspatienten von 44 % auf 31 % – und liegt damit plötzlich gar nicht mehr so weit weg von den Werten der anderen Länder. (7/10)
(Die zweite Zeile seiner Tabelle, in der Schrappe sogar auf einen Anteil von 58 % kommt – und aus denen im Text dann irgendwie sogar 61 % werden – habe ich übrigens nicht beachtet, weil er dabei mit Daten arbeitet, die nach Angaben des RKI noch unvollständig sind.) (9/10)
Ebenfalls schwierig zu erklären, ist die Tatsache, dass laut der Autoren des Thesenpapiers in Deutschland momentan mehr Patienten intensivmedizinisch wegen COVID-19 behandelt werden, als auf Normalstationen. Ein Argument, was die Autoren erstaunlicherweise nicht berücksichtigen, ist die lange Liegezeit (im Schnitt 12-14 Tage, vergleiche hier) von beatmeten COVID-19-Patienten, d.h. dass Patienten aus der „Hochzeit“ der dritten Welle immer noch auf den Intensivstationen liegen, während es mit sinkenden Fallzahlen weniger COVID-19 Patienten mit Normalstationsbehandlungsbedarf gibt.
Kapitel 7: Bettenzahlen: weiterhin fehlende interne Konsistenz
Im siebten Kapitel geht es um die Zahl der von der DIVI gemeldeten Intensivbetten. Und hier setzen sich die Autoren des Thesenpapiers gehörig in die Nesseln.
Es wird nämlich zum Einen eine Diskrepanz in von 2020 von der DIVI gemeldeten Intensivbetten und für den gleichen Zeitraum jetzt gemeldeten Intensivbetten hervorgehoben
Entweder ist in der Vergangenheit aus noch zu klärenden Gründen falsch gezählt worden, oder es hat eine nachträgliche „Korrektur“ der Zahlen gegeben, die ebenfalls erklärungsbedürftig wäre (natürlich ist theoretisch auch ein gezielter Bettenabbau möglich). In jedem Fall stellt sich die Frage, wie es im Nachhinein zu einer Differenz von knapp 3.000 gemeldeten Betten kommen kann, was etwa einem Drittel aller Intensivbetten Frankreichs entspricht oder dem Dreifachen der Kapazität, über die Schweden insgesamt verfügt
der sich aber erstens mit der Herausrechnung der Kinderintensivbetten seit dem 04.03.2021 erklärt, vergleich auch hier und zweitens mit der mittlerweile realistischeren Zählweise der DIVI, die nur noch die auch wirklich betreibbaren Intensivbetten zählt (vergleiche hier und auch hier in der Stellungnahme der DIVI) erklärt.
Zum Anderen kommen die Autoren auf ihren im ersten Kapitel begonnenen Spannungsbogen zurück und führen an, dass ja bislang „nur“ maximal 25 Prozent aller Intensivbetten durch COVID-Patienten belegt wurden. Dadurch sei es unglaubwürdig, dass die COVID-Patienten „alleine“ für die Überlastung auf den Intensivstationen verantwortlich sein. Und dann wird wie folgt argumentiert:
Aktuell werden in vielen Kliniken planbare Eingriffe wieder verschoben, damit es zu keiner Konkurrenz um einen Intensivplatz zwischen diesen Patienten und Corona-Infizierten kommt. Dabei muss man allerdings berücksichtigen, dass die Zahl elektiver Eingriffe in Deutschland im internationalen Vergleich unverhältnismäßig hoch ist.
Und das kann man so nicht stehen lassen. Erstens ist es ja nicht so, dass die COVID-Patienten statt anderer Patienten auf den Intensivstationen liegen (abzüglich der verschobenen elektiven Patienten, dazu aber gleich mehr), sondern sie sind ja on top. Denn anders als in der ersten Welle sehen wir in den Krankenhäusern nicht weniger Herzinfarkt-Patienten usw. Bei den elektiven Patienten fällt die fehlende klinische Erfahrung der Autoren dann endgültig auf. Mit „überflüssigen“ operativen Eingriffen in Deutschland meint man in erster Linie orthopädische Operationen (insbesondere am Knie), Rückenoperationen (vgl. hier) und Herzkatheter-Untersuchungen. Von diesen Patienten muss nur ein verschwindend kleiner Teil postoperativ auf einer Intensivstation behandelt werden. Bei den verschobenen elektiven Operationen mit zu erwartender Intensivpflichtigkeit geht es in einem relevanten Anteil um Tumoroperationen. Und die sind weder überflüssig, noch ambulant leistbar. Alleine bis Sommer 2020 ging die deutsche Krebshilfe von bis zu 50.000 verschobene Tumoroperationen aus (Link). Auch Herzoperationen gehören zu den häufig verschobenen Eingriffen. Für beide Entitäten gilt, dass man sie in einem begrenzten Rahmen nach hinten verschieben kann, aber halt auch nicht unendlich weit. Das heißt, wenn man Rücken- und Knieoperationen und nicht-dringliche Herzkatheter-Eingriffe absagt, ist erst einmal auf den Intensivstationen wenig gewonnen. Außer man macht wilde Personalrochaden und versetzt OP-Personal auf Intensivstationen, was vermutlich weder für die Arbeitszufriedenheit, noch für den zu haltenden qualitativen Standard gut ist. Wobei genau das durch die sogenannten „gemischten Teams“ aus erfahrener Intensivpflege und freiwilligen anzulernenden Pflegekräften aus anderen Arbeitsbereichen ja sogar gemacht wurde.
Kapitel 8: Verfügbarkeit der Intensivkapazität
Im letzten Kapitel geht es um die Verfügbarkeit und Betreibbarkeit von Intensivbetten und um den Personalmangel. Nachvollziehbarer Kritikpunkt ist auch hier die fehlende Datenbasis. Die verfügbaren wenigen Daten deuten hier sogar auf eine Zunahme der Gesamtzahl von Pflegekräften hin. Interessant ist der Punkt, dass die Autoren vermuten, dass eine Erklärung der Abnahme der Intensivbettenzahl seit Sommer 2020 die Wiedereinsetzung der Pflegepersonaluntergrenzen (Link Wikipedia) zum 01. August in der Intensivmedizin. Aus persönlicher Erfahrung würde ich die Vermutung bei diesem Punkt mal streichen, das selbe beschreibt auch die DIVI in ihrer FAQ (Link).
Fazit:
Das Thesenpapier hat Licht- und Schattenseiten. Es weist sehr nachvollziehbar auf Widersprüchlichkeiten und Mängel im Pandemiemonitoring in Deutschland hin. Und den Punkt sehe ich auch als wichtigsten Pluspunkt der Arbeit. Ebenfalls wird der Narrativ der immer jünger werdenden COVID-19-Intensivpatienten entkräftet. Problematisch sind aber in jedem Fall die Fakten- und Recherchefehler, das fehlende Gespür für die Arbeitsrealität in der Intensivmedizin unter Corona-Bedingungen und die Blauäugigkeit zum Thema elektive Operationen, sowie das ständige Beharren auf Ausgleichszahlungen und Investitionspauschalen, die im klinischen Alltag aber keinen interessieren. Ein „DIVIGate“ lässt sich ganz sicher nicht erkennen.
Wo man weiterlesen kann:
Ad hoc-Stellungnahme: Die Pandemie durch SARS-CoV-2/CoViD-19 – Zur intensivmedizinischen Versorgung in der SARS-2/CoViD-19-Epidemie, Link
Michalsen, A., Neitzke, G., Dutzmann, J., Rogge, A., Seidlein, A.-H., Jöbges, S., Burchardi, H., Hartog, C., Nauck, F., Salomon, F., Duttge, G., Michels, G., Knochel, K., Meier, S., Gretenkort, P., & Janssens, U. (2021). Überversorgung in der Intensivmedizin: erkennen, benennen, vermeiden. Medizinische Klinik – Intensivmedizin Und Notfallmedizin, 116(4), 281–294. https://doi.org/10.1007/s00063-021-00794-4





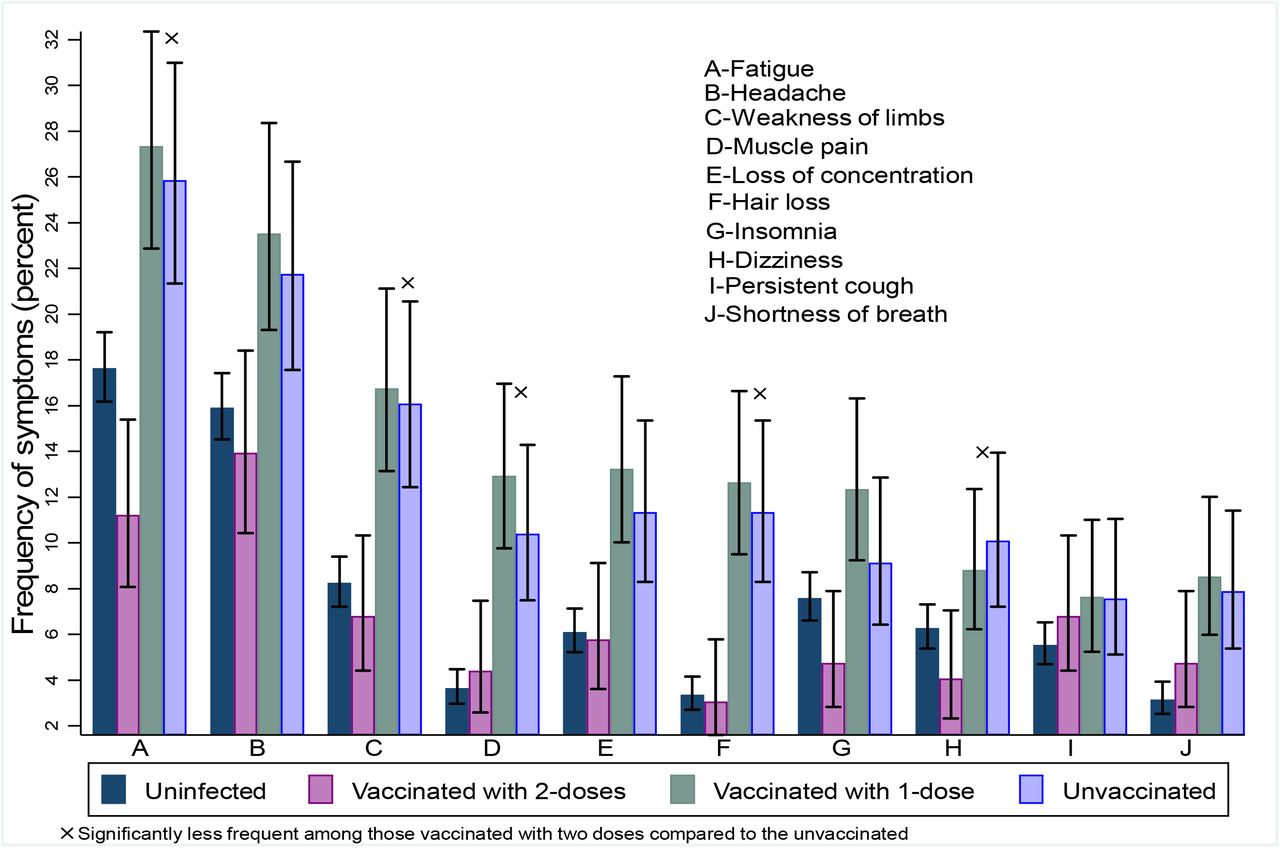{kind=link}